Dilated cardiomyopathy (DCM) is characterized as a disorder of the heart muscle. It is distinguished by the widening/dilation of the left ventricle of the heart with left ventricular or biventricular systolic dysfunction. Contractile functioning of the left ventricle is highly compromised due to dilation. However, in some cases of DCM, the right ventricle is also dilated. Dilated cardiomyopathy (DCM) remains an enigmatic cardiovascular disease (CVD) condition characterized by contractile dysfunction of the myocardium due to dilation of the ventricles. DCM is one of the major forms of CVD contributing to heart failure. Dilation of the left or both ventricles with systolic dysfunction, not explained by known causes, is a hallmark of DCM. Progression of DCM leads to heart failure. Genetic and various other factors greatly contribute to the development of DCM, but the etiology has still remained elusive in a large number of cases.
- dilated cardiomyopathy (DCM)
- heart failure
- gene
- biomarkers
- mechanism
- treatment
- proteomics
- lipidomics
1. Causes of DCM
1.1. Causes of DCM in Children and Newborns
1.1.1. Myocarditis
1.1.2. Selenium Deficiency
1.1.3. Malformation in Pulmonary Arteriovenous
1.1.4. Endocardial Fibroelastosis
1.1.5. Noncompacted Myocardium
1.1.6. Calcium Deficiency
1.1.7. Idiopathic DCM (IDCM)
1.1.8. Barth Syndrome
1.1.9. Familial/Genetic Pediatric DCM
1.2. Causes of DCM in Adults/Adolescents
1.2.1. Familial/Genetic
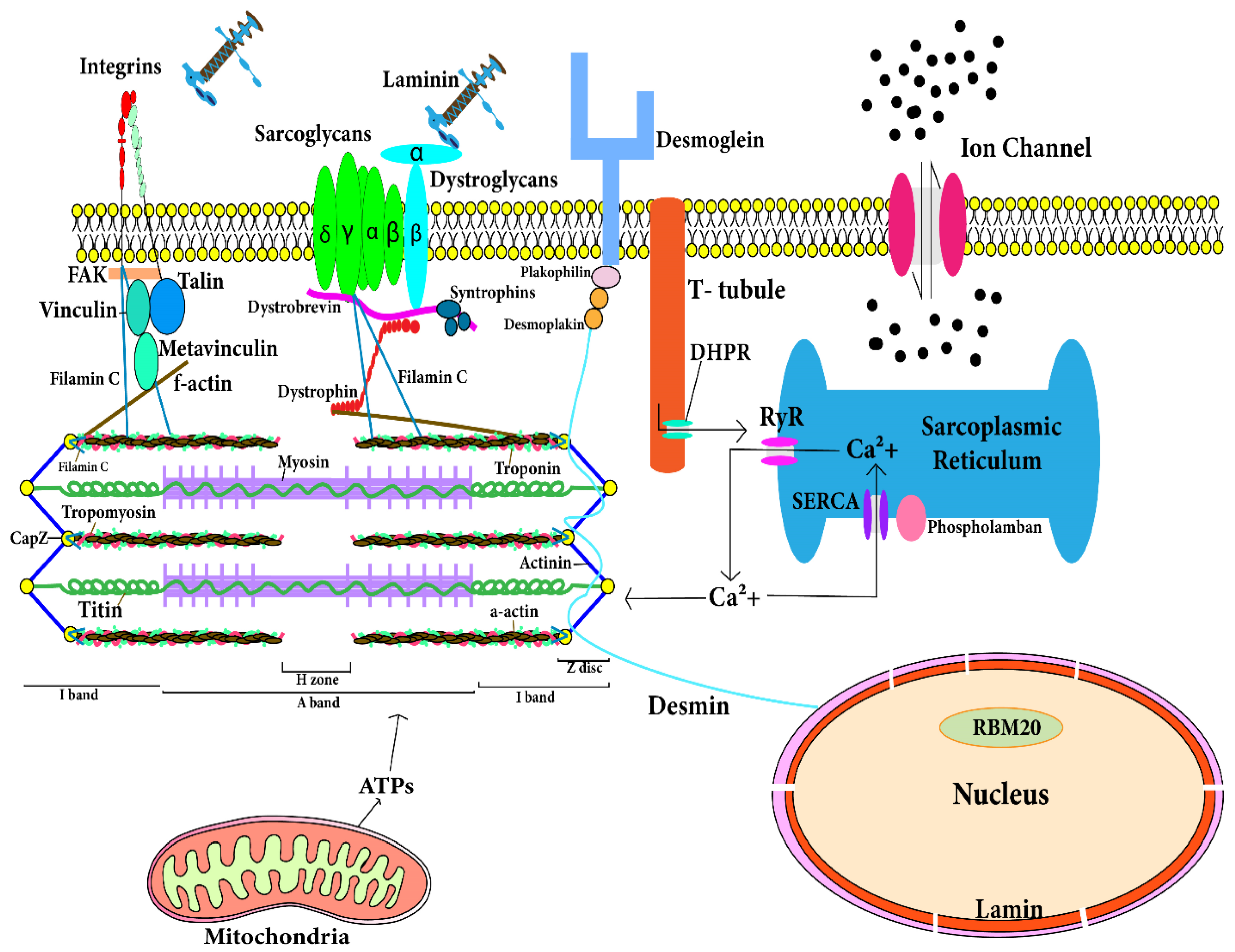
| Cytoskeletal and Sarcomeric Genes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Titin ( | TTN | ) | Cardiac actin alpha | |||||||
| Desmin ( | DES | ) | Troponin T2, I3, C1 ( | TNNT2 | , | TNNI3 | , | TNNC1 | ) | |
| Lamin A/C ( | LMNA | ) | β-myosin heavy chain ( | MHY7 | ) | |||||
| Dilated cardiomyopathy | ventricular remodeling | electrical remodeling |
University of Colorado Anschutz Medical Campus |
α-actinin-2 ( | ACTN2 | ) | Tropomyosin-1 ( | TPM1 | ) | |
| Actin binding LIM domain protein ( | ABLIM1 | ) | Phospholamban ( | PLN | ) | |||||
| Nebulette ( | NEBL | ) | Myosin binding protein C ( | MYBPC3 | ) | |||||
| Myopalladin ( | MYPN | ) | Sodium channel protein type 5 subunit alpha ( | SCN5A | ) | |||||
| Filamin C ( | FLNC | ) | BCL2-associated athanogene 3 ( | BAG3 | ) | |||||
| δ sarcoglycan ( | SGCD | ) | ||||||||
| Vinculin ( | VCL | ) | ||||||||
| Z band alternatively spliced PDZ domain protein ( | ZASP | ) | ||||||||
| Dystrophin gene ( | DMD | ) |
1.2.2. Alcohol
1.2.3. Myocarditis
1.2.4. Tachycardiomyopathy
1.2.5. Mitochondrial Diseases
1.2.6. Cardiomyopathy Associated with Right Ventricular Arrhythmia
1.2.7. Eosinophilic Myocarditis
1.2.8. Toxins
1.2.9. Peripartum Cardiomyopathy
1.2.10. Endocrinopathy
1.2.11. Nutritional Deficiency
2. Diagnosis of DCM
2.1. Clinical Investigation
2.2. Electrocardiography (ECG)
2.3. Echocardiography
2.4. Magnetic Resonance Imaging (MRI)
| Pathological Tests | Other Tests in Specific Indications |
|---|---|
| Erythrocyte sedimentation rate (ESR) | Coronary angiography |
| Viral serology | Blood content tests—carnitine, pyruvate, lactate, autoantibodies, selenium, acylcarnitine profile and drug screening. |
| Creatine kinase (CK) | Red cell transketolase (beri beri) |
| Liver function tests | Urine |
| Renal function | Enteroviruses test |
| Serum ferritin/iron/transferring | Infective screening (HIV/hepatitis C) |
| Thyroid function tests | Organic acid/amino acids |
2.4. Magnetic Resonance Imaging (MRI)
2.5. Serological Test for Virus
2.6. Endomyocardial Biopsy
2.7. Clinical Genetics of DCM
2.8. Pathological Tests for DCM Diagnosis
2.8. Pathological Tests for DCM Diagnosis
| Pathological Tests | Other Tests in Specific Indications |
|---|---|
| Erythrocyte sedimentation rate (ESR) | Coronary angiography |
| Viral serology | Blood content tests—carnitine, pyruvate, lactate, autoantibodies, selenium, acylcarnitine profile and drug screening. |
| Creatine kinase (CK) | Red cell transketolase (beri beri) |
| Liver function tests | Urine |
| Renal function | Enteroviruses test |
| Serum ferritin/iron/transferring | Infective screening (HIV/hepatitis C) |
| Thyroid function tests | Organic acid/amino acids |
2.9.1. Brain Natriuretic Peptide (BNP)
2.9.2. ST2
2.9.3. Troponins T, I
2.9.4. Procollagen Type III
2.9.5. Matrix Metalloproteinase (MMP)
2.9.6. Galectin-3
3. Treatment Strategies of DCM
3.1. Angiotensin-Converting Enzyme (ACE) Inhibitors
3.2. Diuretics
3.3. Angiotensin II (Ang-II) Receptor Antagonists
3.4. Beta Blockers
3.5. Spironolactone
3.6. SGLT2 Inhibitors (SGLT2i)
3.7. Potential Novel Treatments of DCM
3.7.1. Cytokine Antagonists
3.7.2. Anticoagulants
3.7.3. Natriuretic Peptides
3.7.4. Stem Cell Therapy
3.7.5. Clinical Trials
| Drug | Title of Project | Description | Disease or Condition | Location | |||
|---|---|---|---|---|---|---|---|
| ARRY-371797, | ( | p38 inhibitor | ) | A rollover study of ARRY-371797 in patients with LMNA-related dilated cardiomyopathy | Assessment of effectiveness of drug ARRY-371797 is being investigated in this clinical trial. | LMNA-related dilated cardiomyopathy | University of Colorado Aurora, Colorado, United States Johns Hopkins University Baltimore, Maryland, United States |
| ARRY-371797 | A study of ARRY-371797 in patients with symptomatic dilated cardiomyopathy due to a lamin A/C gene mutation | This study is a placebo controlled, dose-dependent efficacy assessment of ARRY-371797 drug on LMNA gene mutation dilated cardiomyopathy patients. | Lamin A/C gene mutation dilated cardiomyopathy | Pfizer Investigational Site Birmingham, Alabama, United States. CB Flock Research Corporation Mobile, Alabama, United States, and 64 more. |
|||
| Ivabradine | Pulse reduction on beta-blocker and Ivabradine therapy | Ivabradine improves ejection fraction by reducing heart rate independently from beta-blockade. | Aurora, Colorado, United States | The Ohio State University Wexner Medical Center Columbus, Ohio, United States |
|||
| Ifetroban | Oral Ifetroban in subjects with Duchenne muscular dystrophy (DMD) | X-linked Duchenne muscular dystrophy (DMD) is a fatal genetic disorder. This lacks effective treatment therapy. Ifetroban is assessed in this study for the treatment of DMD. | Duchenne muscular dystrophy cardiomyopathy dilated cardiomyopathy | Mattel Children’s Hospital Los Angeles, California, United States and 3 more. |
3.8. Assisting Devices and Mechanical Support
3.8.1. Partial Left Ventriculectomy (PLV)
3.8.2. Left Ventricular Assist Devices (LVADs)
3.8.3. Multisite Ventricular Pacing
References
- Caforio, A.L.P.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648.
- Maisch, B.; Richter, A.; Sandmöller, A.; Portig, I.; Pankuweit, S. Inflammatory Dilated Cardiomyopathy (DCMI). Herz 2005, 30, 535–544.
- Kindermann, I.; Kindermann, M.; Kandolf, R.; Klingel, K.; Bültmann, B.; Müller, T.; Lindinger, A.; Boühm, M. Predictors of Outcome in Patients with Suspected Myocarditis. Circulation 2008, 118, 639–648.
- Mahrholdt, H.; Goedecke, C.; Wagner, A.; Meinhardt, G.; Athanasiadis, A.; Vogelsberg, H.; Fritz, P.; Klingel, K.; Kandolf, R.; Sechtem, U. Cardiovascular magnetic resonance assessment of human myocarditis: A comparison to histology and molecular pathology. Circulation 2004, 109, 1250–1258.
- Kearney, M.T.; Cotton, J.; Richardson, P.J.; Shah, A. Viral myocarditis and dilated cardiomyopathy: Mechanisms, manifestations, and management. Postgrad. Med. J. 2001, 77, 4–10.
- Oropeza-Moe, M.; Wisløff, H.; Bernhoft, A. Selenium deficiency associated porcine and human cardiomyopathies. J. Trace Elem. Med. Biol. 2015, 31, 148–156.
- Tirumanisetty, P.; Abdullah, A.; Matos, M. Pulmonary arteriovenous malformation: A myriad of presentations. Chest 2018, 154, 1032A.
- Seki, A.; Patel, S.; Ashraf, S.; Perens, G.; Fishbein, M.C. Primary endocardial fibroelastosis: An underappreciated cause of car-diomyopathy in children. Cardiovasc. Pathol. 2013, 22, 345–350.
- Yin, L. Non-compact cardiomyopathy or ventricular non-compact syndrome? J. Cardiovasc. Ultrasound 2014, 22, 165–172.
- Sanyal, D.; Raychaudhuri, M. Infants with dilated cardiomyopathy and hypocalcemia. Indian J. Endocrinol. Metab. 2013, 17, 221–223.
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardio-myopathy in children. Jama 2006, 296, 1867–1876.
- Jefferies, J.L. Barth syndrome. Am. J. Med. Genet. Part C (Semin. Med. Genet.) 2013, 163, 198–205.
- Aprikyan, A.A.; Khuchua, Z. Advances in the understanding of Barth syndrome. Br. J. Haematol. 2013, 161, 330–338.
- Rampersaud, E.; Siegfried, J.D.; Norton, N.; Li, D.; Martin, E.; Hershberger, R.E. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog. Pediatr. Cardiol. 2011, 31, 39–47.
- Khan, R.S.; Pahl, E.; Dellefave-Castillo, L.; Rychlik, K.; Ing, A.; Yap, K.L.; Brew, C.; Johnston, J.R.; McNally, E.M.; Webster, G. Genotype and Cardiac Outcomes in Pediatric Dilated Cardiomyopathy. J. Am. Hear. Assoc. 2022, 11, e022854.
- Luisa, M.; Francesca, B.; Anita, S.; Gianfranco, S.; Taylor, M.R. Genetic causes of Dilated Cardiomyopathy. Prog. Pediatr. Cardiol. 2014, 37, 13–18.
- Arbustini, E.; Narula, N.; Dec, G.W.; Reddy, K.S.; Greenberg, B.; Kushwaha, S.; Marwick, T.; Pinney, S.; Bellazzi, R.; Favalli, V.; et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: Endorsed by the World Heart Federation. J. Am. Coll. Cardiol. 2013, 62, 2046–2072.
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32.
- Hershberger, R.E.; Cowan, J.; Jordan, E.; Kinnamon, D.D. The Complex and Diverse Genetic Architecture of Dilated Cardiomyopathy. Circ. Res. 2021, 128, 1514–1532.
- Maisch, B. Alcoholic cardiomyopathy: The result of dosage and individual predisposition. Herz 2016, 41, 484–493.
- Mohamed, H.A. Tachycardia-induced Cardiomyopathy (Tachycardiomyopathy). Libyan J. Med. 2007, 2, 26–29.
- Bates, M.G.; Bourke, J.P.; Giordano, C.; D’Amati, G.; Turnbull, D.M.; Taylor, R.W. Cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur. Heart J. 2012, 33, 3023–3033.
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300.
- Bluett, R.; McDonnell, D.; O’Dowling, C.; Vaughan, C. Eosinophilic myocarditis as a first presentation of eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). Case Rep. 2017, 2017, bcr-2017.
- Nakayama, T.; Murai, S.; Ohte, N. Dilated Cardiomyopathy with Eosinophilic Granulomatosis with Polyangiitis in Which Active Myocardial Inflammation Was Only Detected by Endomyocardial Biopsy. Intern. Med. 2018, 57, 2675–2679.
- Chen, M.-X.; Yu, B.-L.; Peng, D.-Q.; Zhou, S.-H. Eosinophilic myocarditis due to Churg–Strauss syndrome mimicking reversible dilated cardiomyopathy. Hear Lung 2014, 43, 45–47.
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Clas-sification and Diagnosis: A Scientific Statement from the American Heart Association. Circulation 2019, 140, e9–e68.
- Pearson, G.D.; Veille, J.C.; Rahimtoola, S.; Hsia, J.; Oakley, C.M.; Hosenpud, J.D.; Ansari, A.; Baughman, K.L. Peripartum cardiomyopathy: National Heart, Lung, and Blood Institute and Office of Rare Diseases (National Institutes of Health) workshop recommendations and review. Jama 2000, 283, 1183–1188.
- Klein, I.; Danzi, S. Thyroid disease and the heart. Circulation 2007, 116, 1725–1735.
- Mobine, H.R.; Baker, A.B.; Wang, L.; Wakimoto, H.; Jacobsen, K.C.; Seidman, C.E.; Seidman, J.G.; Edelman, E.R. Pheochromocytoma-induced cardiomyopathy is modulated by the synergistic effects of cell-secreted factors. Circ. Heart Fail. 2009, 2, 121–128.
- Bradley, T.J.; Slorach, C.; Mahmud, F.H.; Dunger, D.B.; Deanfield, J.; Deda, L.; Elia, Y.; Har, R.L.H.; Hui, W.; Moineddin, R.; et al. Early changes in cardiovascular structure and function in adolescents with type 1 diabetes. Cardiovasc. Diabetol. 2016, 15, 31.
- Albakri, A. Nutritional deficiency cardiomyopathy: A review and pooled analysis of pathophysiology, diagnosis and clinical management. Res. Rev. Insights 2019, 3, 1–19.
- Dilated Cardiomyopathy (DCM)-Cardiomyopathy UK . Available online: https://www.cardiomyopathy.org/dilated-cardiomyopathy/intro (accessed on 20 April 2021).
- Ku, L.; Feiger, J.; Taylor, M.; Mestroni, L. Familial Dilated Cardiomyopathy. Circulation 2003, 108, e118–e121.
- Duran, J.; Martinez, A.; Adler, E. Cardiovascular Manifestations of Mitochondrial Disease. Biology 2019, 8, 34.
- Dilated Cardiomyopathy (DCM) • LITFL • ECG Library Diagnosis . Available online: https://litfl.com/dilated-cardiomyopathy-dcm-ecg-library/ (accessed on 20 April 2021).
- Finocchiaro, G.; Merlo, M.; Sheikh, N.; De Angelis, G.; Papadakis, M.; Olivotto, I.; Rapezzi, C.; Carr-White, G.; Sharma, S.; Mestroni, L.; et al. The electrocardiogram in the diagnosis and management of patients with dilated cardiomyopathy. Eur. J. Hear Fail. 2020, 22, 1097–1107.
- Mathew, T.; Williams, L.; Navaratnam, G.; Rana, B.; Wheeler, R.; Collins, K.; Harkness, A.; Jones, R.; Knight, D.; O’Gallagher, K.; et al. Diagnosis and assessment of dilated cardio-myopathy: A guideline protocol from the British Society of Echocardiography. Echo Res. Pract. 2017, 4, G1–G13.
- Green, J.J.; Berger, J.S.; Kramer, C.M.; Salerno, M. Prognostic Value of Late Gadolinium Enhancement in Clinical Outcomes for Hypertrophic Cardiomyopathy. JACC Cardiovasc. Imaging 2012, 5, 370–377.
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010.
- Wu, L.A.; Lapeyre, A.C.; Cooper, L.T. Current Role of Endomyocardial Biopsy in the Management of Dilated Cardiomyopathy and Myocarditis. Mayo Clin. Proc. 2001, 76, 1030–1038.
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19.
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547.
- Twu, C.; Liu, N.Q.; Popik, W.; Bukrinsky, M.; Sayre, J.; Roberts, J.; Rania, S.; Bramhandam, V.; Roos, K.P.; MacLellan, W.R.; et al. Cardiomyocytes undergo apoptosis in human immunode-ficiency virus cardiomyopathy through mitochondrion and death receptor-controlled pathways. Proc. Natl. Acad. Sci. USA 2002, 99, 14386–14391. Available online: www.pnas.org (accessed on 21 April 2021).
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5346598/ (accessed on 21 April 2021).
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.P.; Charron, P.; Blanes, J.G.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Hear J. 2012, 34, 1448–1458.
- Suresh, C.P.; Saha, A.; Kaur, M.; Kumar, R.; Dubey, N.K.; Basak, T.; Tanwar, V.S.; Bhardwaj, G.; Sengupta, S.; Batra, V.V.; et al. Differentially expressed urinary biomarkers in children with idiopathic nephrotic syndrome. Clin. Exp. Nephrol. 2015, 20, 273–283.
- Felker, G.M.; Hasselblad, V.; Hernandez, A.F.; O’Connor, C.M. Biomarker-guided therapy in chronic heart failure: A meta-analysis of randomized controlled trials. Am. Hear J. 2009, 158, 422–430.
- Januzzi, J.L.; Peacock, W.F.; Maisel, A.S.; Chae, C.U.; Jesse, R.L.; Baggish, A.L.; O’Donoghue, M.; Sakhuja, R.; Chen, A.A.; van Kimmenade, R.R.; et al. Measurement of the Interleukin Family Member ST2 in Patients with Acute Dyspnea: Results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) Study. J. Am. Coll. Cardiol. 2007, 50, 607–613.
- DeFilippi, C.R.; de Lemos, J.A.; Christenson, R.H.; Gottdiener, J.S.; Kop, W.J.; Zhan, M.; Seliger, S.L. Association of serial measures of cardiac troponin T using a sensitive assay with incident heart failure and cardiovascular mortality in older adults. Jama 2010, 304, 2494–2502.
- Cicoira, M.; Rossi, A.; Bonapace, S.; Zanolla, L.; Golia, G.; Franceschini, L.; Caruso, B.; Marino, P.N.; Zardini, P. Independent and additional prognostic value of aminoterminal propeptide of type III procollagen circulating levels in patients with chronic heart failure. J. Card. Fail. 2004, 10, 403–411.
- Yan, A.T.; Yan, R.T.; Spinale, F.G.; Afzal, R.; Gunasinghe, H.R.; Arnold, M.; Demers, C.; Mckelvie, R.S.; Liu, P.P. Plasma Matrix Metalloproteinase-9 Level Is Correlated with Left Ventricular Volumes and Ejection Fraction in Patients with Heart Failure. J. Card. Fail. 2006, 12, 514–519.
- Kapur, N.K.; Heffernan, K.S.; Yunis, A.A.; Parpos, P.; Kiernan, M.S.; Sahasrabudhe, N.A.; Kimmelstiel, C.D.; Kass, D.A.; Karas, R.H.; Mendelsohn, M.E. Usefulness of Soluble Endoglin as a Noninvasive Measure of Left Ventricular Filling Pressure in Heart Failure. Am. J. Cardiol. 2010, 106, 1770–1776.
- Herman, L.L.; Padala, S.A.; Annamaraju, P.; Bashir, K. Angiotensin Converting Enzyme Inhibitors (ACEI); StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Packer, M.; Poole-Wilson, P.A.; Armstrong, P.W.; Cleland, J.G.; Horowitz, J.D.; Massie, B.M.; Rydén, L.; Thygesen, K.; Uretsky, B.F. Comparative effects of low and high doses of the angiotensin-converting enzyme inhibitor, lisinopril, on morbidity and mortality in chronic heart failure. ATLAS Study Group. Circulation 1999, 100, 2312–2318.
- Diuretics . Available online: https://www.cvpharmacology.com/diuretic/diuretics (accessed on 19 April 2021).
- Ballew, J.R.; Fink, G.D. Characterization of the antihypertensive effect of a thiazide diuretic in angiotensin II-induced hy-pertension. J. Hypertens. 2001, 19, 1601–1606.
- Barreras, A.; Gurk-Turner, C. Angiotensin Ii Receptor Blockers. Bayl. Univ. Med Cent. Proc. 2003, 16, 123–126.
- Beta-Blockers for Heart Disease: Benefits, Risks, and More . Available online: https://www.healthline.com/health/heart-disease/beta-blockers (accessed on 19 April 2021).
- Böhm, M.; Maack, C. Treatment of heart failure with beta-blockers. Mechanisms and results. Basic Res. Cardiol. 2000, 95, I15–I24.
- James, P.A.; Oparil, S.; Carter, B.L.; Cushman, W.C.; Dennison-Himmelfarb, C.; Handler, J.; Lackland, D.T.; LeFevre, M.L.; MacKenzie, T.D.; Ogedegbe, O.; et al. 2014 evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). Jama 2014, 311, 507–520.
- Sica, D.A. Pharmacokinetics and Pharmacodynamics of Mineralocorticoid Blocking Agents and their Effects on Potassium Homeostasis. Hear Fail. Rev. 2005, 10, 23–29.
- Miller, E.; Shubrook, J.H. Sodium glucose co-transporter 2 inhibitors in the treatment of type 2 diabetes mellitus. Am. Coll. Osteopath. Fam. Physicians 2015, 7, 10–30.
- Zou, H.; Zhou, B.; Xu, G. SGLT2 inhibitors: A novel choice for the combination therapy in diabetic kidney disease. Cardiovasc. Diabetol. 2017, 16, 65.
- Blum, A.; Miller, H. Pathophysiological role of cytokines in congestive heart failure. Annu. Rev. Med. 2001, 52, 15–27.
- Hassan, I.; Dorjay, K.; Anwar, P. Pentoxifylline and its applications in dermatology. Indian Dermatol. Online J. 2014, 5, 510–516.
- Rubin, L.J.; Roux, S. Bosentan: A dual endothelin receptor antagonist. Expert Opin. Investig. Drugs 2002, 11, 991–1002.
- Koniaris, L.S.; Goldhaber, S.Z. Anticoagulation in dilated cardiomyopathy. J. Am. Coll. Cardi-Ology 1998, 31, 745–748.
- Riegger, A.J.G. Atrial Natriuretic Peptide in Heart Failure. In Endocrinology of the Heart; Springer: Berlin, Heidelberg, 1989; pp. 85–89.
- Seth, S.; Bhargava, B.; Narang, R.; Ray, R.; Mohanty, S.; Gulati, G.; Kumar, L.; Airan, B.; Venugopal, P. The ABCD (Autologous Bone Marrow Cells in Dilated Cardiomyopathy) Trial. J. Am. Coll. Cardiol. 2010, 55, 1643–1644.
- U.S. National Library of Medicine. Clinical Trials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 17 September 2020).
- Yoshii, S.; Hosaka, S.; Suzuki, S.; Osawa, H.; Akashi, O.; Tada, Y.; Sugiyama, H.; Hoshiai, M.; Tan, T.; Kadono, T.; et al. Indications for partial left ventriculectomy in pediatric dilated cardiomyopathy. Circ. J. 2002, 66, 337–340.
- Franco-Cereceda, A.; McCarthy, P.M.; Blackstone, E.H.; Hoercher, K.J.; White, J.A.; Young, J.B.; Starling, R.C. Partial left ventriculectomy for dilated cardiomyopathy: Is this an alternative to transplantation? J. Thorac. Cardiovasc. Surg. 2001, 121, 879–893.
- Mancini, D.; Colombo, P.C. Left ventricular assist devices: A rapidly evolving alternative to transplant. J. Am. Coll. Cardiol. 2015, 65, 2542–2555.
- Sipahi, I.; Fang, J.C. QRS duration criteria to select patients for cardiac resynchronization therapy: Crt should be reserved for a QRS duration ≥150 ms: Pro. Circ. Arrhythmia Electrophysiol. 2013, 6, 436–441.