Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Alessandra Gallo.
Voltage-gated ion channels are plasma membrane proteins that generate electrical signals following a change in the membrane voltage.
- conotoxins
- voltage-gated ion currents
- sodium
1. Introduction
Marine organisms produce a great variety of toxins. Among them, venomous mollusks cone snails have provided so far more than 6000 different toxins that have been isolated and characterized by more than 100 different species. Cone snails have been the object of huge interest and study since ancient times due to their shell features but mostly for their poison-associated toxins. The main physiological roles of these toxins are for self-defense from predators but also to predate themselves and to compete with other marine species, also thanks to a poisonous sting that may be fatal for humans. The venomous properties of the toxins have been revealed to exert pharmacological bioactivities, especially on a vast variety of pain-associated neurological disorders. Hence, the toxin’s level of poisonousness has been used as an efficient and beneficial pharmacological and therapeutic tool, making conids good candidates for new drug design and development [1,2,3][1][2][3]. Conus-derived toxins, known as conotoxins (CnTX), are venomous small peptides consisting of 5–50 amino acid residues with multiple disulfide bonds among cysteines. An old classification divided CnTX into cysteine-rich and cysteine-poor groups. At present, instead, three CnTX groups are classified based on the cysteine framework gene superfamily and the pharmacological family.
Currently, pharmacological family classification is related to the receptor target and the type of CnTX-target interaction. CnTX included in the same superfamily share a similar signal peptide sequence, which undergoes structural and functional differentiation when they become encoded mature peptides [4,5][4][5]. Nonetheless, over 10,000 CnTX sequences have been disclosed and published; however, 3D structural and functional information is still lacking and their pharmacological characterizations have not been elucidated. CnTX includes several different pharmacological families that selectively target specific voltage-gated ion channels, G protein-coupled receptors, enzymes, and transporters [6,7,8][6][7][8]. Currently, CnTXs are under evaluation by neuroscientists and drug developers for their peculiar selectivity to mammalian and human targets and, in particular, for their ability to inhibit voltage-gated ion channels. As an example, the µ-CnTX family exerts its activity by combining multiple peptides action known as “cabal”, which is aimed to ensure the most effective bioactivity through ion channels modulation. Due to their ability to interact with human ion channels, these toxins are considered “specialists in neuropharmacology” and given their therapeutic potential, some of them have been involved in human clinical treatments [9,10,11][9][10][11]. Based on their specific selectivity, CnTX represent basic tools to elucidate ion channel function and their involvement in biological mechanisms and processes. In this revisew, wearch, the researchers focus on the main CnTX pharmacological properties and their modulation of voltage-gated sodium (NaV), calcium (CaV), and potassium (KV) channels acting through different and, sometimes, opposite physiological and pathological mechanisms (Figure 1).
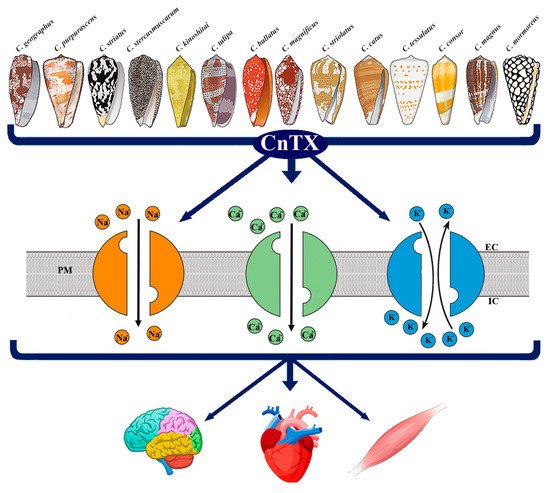
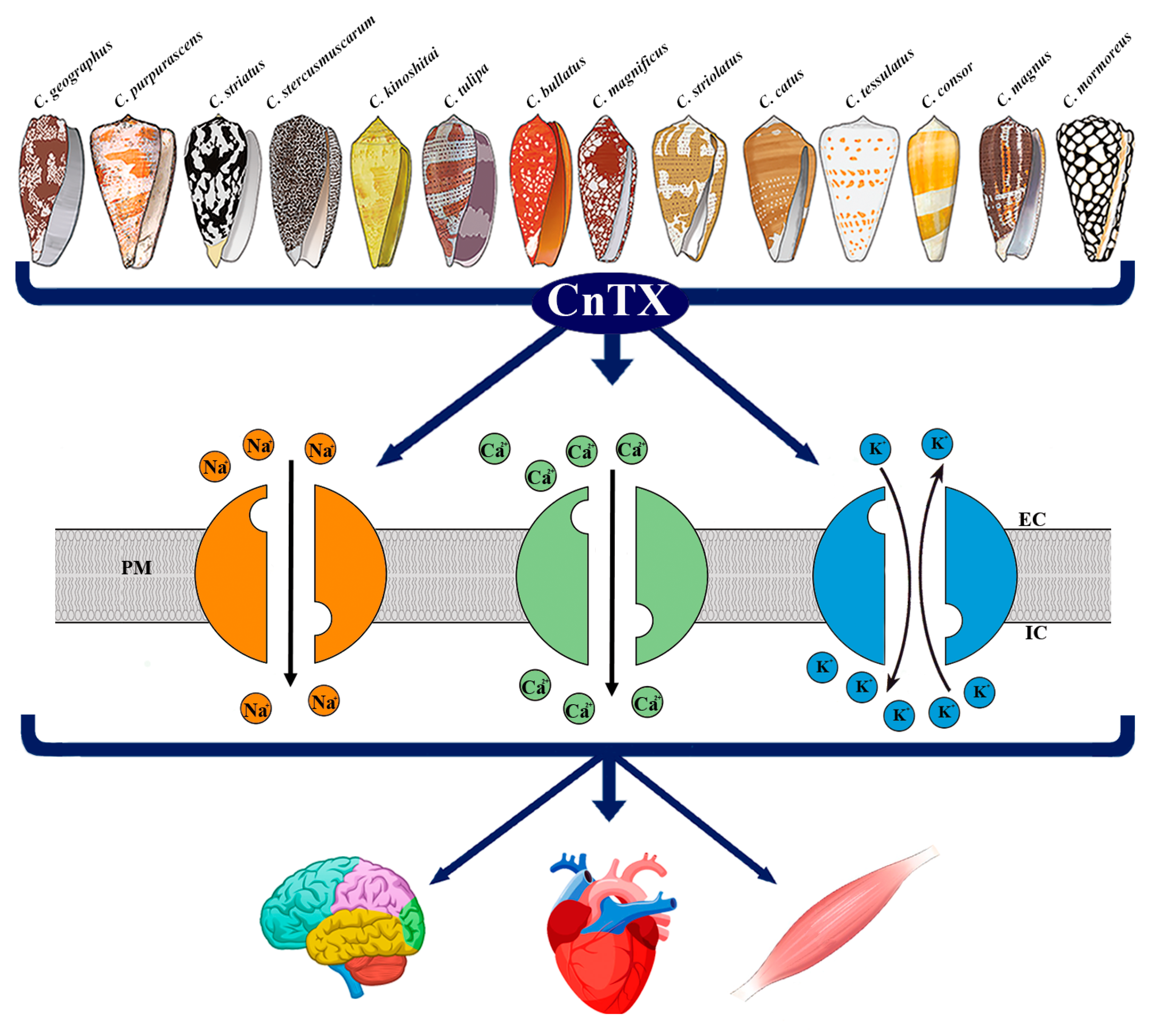 Figure 1. Representative image of CnTX bioactivity via voltage-gated ion channel modulation. Conus derived toxins (CnTX) target numerous and different NaV and/or CaV and/or KV channel subtypes generating ion current fluxes through neurons of central and peripheral nervous systems as well as in heart and skeletal muscle cells. IC = intracellular compartment; EC = extracellular compartment; PM = plasma membrane.
Figure 1. Representative image of CnTX bioactivity via voltage-gated ion channel modulation. Conus derived toxins (CnTX) target numerous and different NaV and/or CaV and/or KV channel subtypes generating ion current fluxes through neurons of central and peripheral nervous systems as well as in heart and skeletal muscle cells. IC = intracellular compartment; EC = extracellular compartment; PM = plasma membrane.2. Pathophysiological Response to CnTX Voltage-Gated Channel Modulation
2.1. Na
V
Channels
NaV channels are voltage-gated ion channels responsible for the generation of the rapid RP depolarization known as action potential that, in excitable cells, propagates electrical signals in muscles and nerves in either the central or peripheral nervous system [33][12]. Since NaV channels underlie neurotransmission, contraction, excitation coupling, and associated physiological functions [33][12], mutations and defects in their functional activity are associated with several neurological disturbances and channelopathies [34][13]. From a structural point of view, NaV channels are heteromeric complexes with a pore-forming α subunit of about 260 KDa linked to one or two β subunits of different molecular weights. The α subunit contains the binding site for several neurotoxins and drugs that target the channel and significantly change its activity. The β subunits are involved in different signaling roles in physiological processes such as cell adhesion, gene regulation, and brain development and in the kinetic regulation of channel opening. Both α and β subunits contain the receptors for toxins targeting the channel. Currently, characterized according to the α-pore-forming subunit sequences, nine isoforms of the NaV channels α subunits (1.1–1.9) have been identified. Specifically, isoforms 1.1, 1.2, 1.3, and 1.6 are predominantly expressed in the central nervous system, whereas isoforms 1.7, 1.8, and 1.9 are mainly expressed in the peripheral nervous system. In addition, skeletal and heart muscles contain 1.4 and the 1.5 isoforms, respectively [35,36,37,38][14][15][16][17].
Toxins and venom compounds targeting NaV channels are of particular importance due to their pivotal role played in the neuromuscular system. Together with their pathophysiological action, CnTX have also provided basic information on the molecular structure, function, and subtype-selectivity of NaV channels [39][18] (Table 1).
Table 1.
CnTX subfamilies targeting voltage-gated sodium (Na
V
) channel subtypes, functional impact, and pathophysiological activity.
| Species | CnTX Subfamilies | Channel Subunit Targeted | Functional Impact | Pathophysiological Activity | References | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C. geographus | C. striatus | μ-GIIIA | Na | V | 1.4 | block skeletal muscle channels | paralysis | [40] | [19] | ||||||||||||||||||
| kA-SIVA | K | V | block | spastic paralytic symptoms | [98] | [75] | C. geographus | μ-GIIIB μ-GIIIC |
Na | V | 1.1 Na | V | 1.2 Na | V | 1.4 Na | V | 1.6 | discriminate between muscle and neuronal channels | - | [41 | |||||||
| C. purpurascens | K-PVIIA | ] | K | V | 1.3 | inhibition | therapeutics for multiple sclerosis, rheumatoid arthritis, diabetes, and dermatitis[20] |
||||||||||||||||||||
| [ | 99 | , | 100 | ] | [76][77] | C. bullatus | C. catus | C. consor | C. magnus | C. purpurascens | C. stercusmuscarum | C. striatus | C.tulipa | μ-CnIIIA μ-CnIIIB μ-CnIIIC μ-CIIIA, μ-MIIIA |
Na | V | 1 | block channel conductance | paralysis (CIIIA) | [42] | |||||||
| C. radiatus | kM-RIIIK K-CnTX RIIIJ |
Human K | V | 1.2 K | V | 1.2–K | V | 1.5 | [43] | [21] |
block target[22] |
||||||||||||||||
| cardio-protective action | no activity | [ | 101 | ,102] | [78][79] | C. purpurascens | μ-PIIIA | Na | V | 1.2 Na | V | 1.4 Na | V | 1.7 | inhibit channel modulation | - | [44] | [23] | |||||||||
| C. stercusmuscarum | μ-SmIIIA | irreversible block of NaV currents |
nociceptive role | [45,46] | [24] |
]. Isolated from the venom of Conus stercusmuscarum, μ-SmIIIA, due to its strong affinity for neuronal NaV channel subtypes, it can irreversibly block NaV currents in different neurons of either amphibians or rats, demonstrating a nociceptive role [45,46][24][25].
2.2. Ca
V
Channels
Ca2+ is the signaling ion for which its elevation from the resting state is involved in many physiologic processes in most cell types. Ca2+ homeostasis is regulated by an intricate connection of intracellular organelles, binding molecules, and transmembrane channels and transporters. Rapid Ca2+ entry into the neurons gives rise to action potential propagation along cells, activating a cascade of processes such as enzyme activation and gene regulation. CaV channels are at the origin of depolarization evoked by Ca2+ entry into excitable cells of either brain or muscle tissues. This, in turn, is responsible for most physiological functions as Ca2+ dependent muscle contraction, neurotransmitters release, gene transcription, and others. Dysfunctions in these processes, therefore, may alter neurotransmission and gene transcription generating neuropathic pain and related disease states [63,64][40][41]. Ca2+ channels are made by 4–5 different subunits, among which the α1 includes the voltage sensor, related apparatus, and the conduction pore [65][42]. According to the voltage changes and depolarization amplitude needed for their activation, CaV channels are organized into two categories: high- and low-voltage activated Ca2+ channels. The most known members of CaV channels are (i) the high voltage-activated L-type characterized by slow voltage-dependent inactivation involved in cell excitability, contraction, gene expression regulation, and oocyte maturation; and (ii) the P/Q-, N-, and R-types that are more prominently active in fast neuronal signal transmissions [23,66][43][44]. T-types are the low voltage-activated channels present in either neurons or smooth and cardiac muscular tissues. N-type CaV2.1 and CaV2.2, in particular, play important roles in the transmission of pain signals to the central nervous system. About ten different genes encode for different types of CaV subunits that are grouped into three major classes (CaV1, CaV2, and CaV3). Specifically, the CaV1 family encodes four different types of L-type channels, CaV2 family for 2.1, 2.2, and 2.3 corresponding to P/Q type, N-type, and R-type channels, respectively, whereas the CaV3 family includes three different types of T-type CaV channels [67][45]. Following channel gating, once Ca2+ ions are released into the cytosol, they behave as second messengers binding a large number of proteins and, in turn, influence multiple cell functions and complete several physiological processes [68][46]. Although most of the studies have focused on the pivotal involvement of CaV1.2 and CaV2.2 isoforms in the modulation of pain states, some hints also revealed the involvement of T-type CaV3.1–CaV3.3 with a special focus on the CaV3.2 knockdown effect in mechanical, thermal, and chemical pain diseases [69][47] (Table 2).
Table 2.
CnTX subfamilies targeting voltage-gated calcium (Ca
V
) channel subtypes, functional impact, and pathophysiological activity.
| Species | CnTX Subfamilies | Channel Subunit Targeted | Functional Impact | Pathophysiological Activity | References | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C. pennaceus | ω-PnVIA ω-PVIB |
HVA Ca | V | selectively but reversibly block HVA currents | - | [70] | [48] | |||||||||||||||
| C. textile | ω-TxVII | Ca | V | block Ca | V | currents | - | [71] | [49] | |||||||||||||
| C. geographus | ω-GVIA | Ca | V | irreversibly block Ca | V | channels | - | [72] | [50] | |||||||||||||
| C. magnus | ω-MVIIA ω-MVIIC |
|||||||||||||||||||||
| C. striatus | K-Conk-S1; K-Conk-S2 | Ca | V | 2.2 P/Q-type Ca | V | 2.1 and Ca | V | 2.2 | K | V | 1.7inhibits channel activity blocks channel activity |
targetanalgesic on chronic pain neuroprotective effect |
therapeutics for diabetes | [73,74] | [51][52] | |||||||
| [ | 103 | ] | [ | 80 | ] | C. moncuri | ω-MoVIA ω-MoVIB |
|||||||||||||||
| C. capitaneus | C. miles | C. vexillum | C. striatus | Ca | V | 2.2 | channel affinity | - | C.imperialis[75 | I-superfamily conus peptides | K | V | 1.1 K | V | 1.3 | ] | [ | block[ | -25] | |||
| [ | 104 | , | C. striatus | μ-SIIIA | Na | V | 1.2 | block of neuronal NaV current |
analgesic activity | [47] | [26] | |||||||||||
| C. tulipa | C. kinoshitai | C. striatus | μ-TIIIA, μ-KIIIA, μ-KIIIB, μ-SIIIB |
Na | V | 1.1 Na | V | 1.2 Na | V | 1.3 Na | V | 1.4 Na | V | 1.6 | affinity Na | V | channels | analgesic activity | [48,49,50] | [27][28][29] | ||
| C. marmoreus | μO-MrVIA, μO MrVIB, μO MfVIA |
Na | V | 1.8 | inhibit channel activity | analgesic activity | [51] | [30] | ||||||||||||||
| C. radiatus | ί-RXIA | Na | V | 1.6 | shift channel activation | - | [52] | [31] |
Several CnTX families and, in particular, µ-, µO-, δ-, and ι′-CnTX can target NaV channels upon binding specific sites on the α subunits. In particular, they may act as pore blockers that structurally occlude the pore or interfere with voltage sensors [53,54][32][33]. These families, in fact, differently modulate channel activity acting as either inhibitors (µ- and µO-CnTX) or stimulators (δ- and ι′-CnTX) of NaV activity. The channel inhibition is underlined by different mechanisms. For example, µ-CnTX blocks ion conductance by binding to the channel’s external vestibule, whereas µO-CnTXs are gating modulators that bind the external side of the pore, causing channel closure [55,56][34][35]. Similarly, δ- and ι′-CnTX activate channels by two different mechanisms that prolong the channel’s opening and shift voltage activation to more hyperpolarized potentials, respectively [57][36]. µ-CnTX are small peptides composed of 16–22 amino acids and three disulfide bonds that, by inhibiting the α-subunit of NaV channels, affect neuromuscular transmission causing paralytic to analgesic effects in mammals [58,59][37][38]. Currently, among the 12 CnTX known, μGIIIA was the first one to be isolated from Conus geographus; by binding the NaV1.4 sub-type channel pore, it exerts the inhibition of rat skeletal muscle channels [40][19]. From the same venom, the two isoforms μ-GIIIB and μ-GIIIC, differing for only four residues, exhibited a different affinity for the muscle subtype NaV channels 1.4 and the neuronal NaV1.1, NaV1.2, and NaV1.6 channel subtypes. These μCnTX can discriminate between muscle and neuronal NaV channels, becoming candidates for therapeutic plans on neurological disorders. Therefore, a wide range of studies was aimed to identify whether interactions between several μ-CnTX and NaV channel subtypes may act as selective inhibitors of NaV channels neuronal subtypes, with possible clinical impact on neurological processes [41][20]. In this light, several μ-CnTXs with affinities for neuronal NaV channel subtypes were isolated from Conus bullatus, catus, consor, kinoshitai, magnus, purpurascens, stercusmuscarum, striatus, striolatus, and tulipa. Among these, μ-CnIIIA, μ-CnIIIB, μ-CnIIIC, μ-CIIIA, and μ-MIIIA can block the conductance of NaV1 subtypes in amphibian neurons. Some of them, by inhibiting olfactory and sciatic nerve action potentials, modulate pain signals and exert analgesic activity [42][21]; however, only CIIIA may cause paralysis [43][22]. μ-PIIIA is a peculiar versatile μ-CnTX that can differentiate NaV channel subtype isoforms by inhibiting NaV1.2, NaV1.4, and NaV1.7 channel subtypes from rat brain, skeletal muscle, and peripheral nerves, respectively. Furthermore, recent studies revealed that μ-PIIIA is a strong inhibitor of NaV channels involved in muscle contraction and, specifically, of NaV neuronal subtypes in both the central and peripheral nervous systems [44,60][23][39
| 53 | ||||||||||||||
| ] | ||||||||||||||
| 105 | ||||||||||||||
| ] | ||||||||||||||
| [ | 81 | ] | [ | 82 | ] | |||||||||
| C. striatus | ω-SVIA ω-SVIB ω-SO-3 |
Ca | V | 2.2 Ca | V | 2.1 and Ca | V | 2.2 N-type Ca | V | 2.2 | targeting binding affinity inhibition |
|||
| C. virgo | ViTx | K | V | 1.1 K | V | 1.3 | inhibitionparalytic effect lethal injection attenuates acute and chronic pain |
- | [76,77] | [54][55] | ||||
| [ | 106 | ] | [ | 83 | ] | C. catus | ω-CVIE ω-CVIF ω-CVID |
Ca | V | N-type Ca | V | 2.2 | affinity antagonist activity | inhibition of nociceptive pain; reducing allodynic behaviour alleviates chronic neuropathic pain reduce allodynic behaviour |
| C. purpurescens | CGX-1051 | K | V | [ | 78,79,80] | [56][57][58] | ||||||||
| C. fulmen | ω-FVIA | N-type Ca | V | 2.2 | inhibition | reduces nociceptive behaviour, neuropathic pain, mechanical and thermal allodynia | [81] | [59] | ||||||
| C. textile | ω-CNVIIA | N-type Ca | V | 2.2 | inhibition | blocks neuromuscular junction, paralysis, death | [82] | [60] | ||||||
| C. pergrandis | α-PeIA | GABAB receptors coupled to N-type Ca | V | blocking activity | analgesic activity | [83] | [61] | |||||||
| C. victoriae | C. regius | α-Vc1.1 α-RgIA α-AuIB α-MII |
GABAB receptors coupled to N-type Ca | V | 2.2. | inhibition | analgesic activity on sciatic nerve ligation injury; allodynia relieves | [84,85,86,87] | [62][63][64][65] |
In the 1990s, numerous studies aimed at identifying new conopeptides, which which its homologues would exhibit possible affinity for CaV channels. However, it was only later that these peptides were characterized and their possible similarities disclosed. The in vitro studies of toxin administration and, in particular, their effects on neuropathic disturbs have been progressively evidenced, and the clinical applications for relieving different pathologies were established. The most known CnTX able to modulate CaV channels, by occluding the channel pore and, thus, preventing Ca2+ entry, is the ω-CnTX family. Typically, ω-CnTX are peptides that are composed of 24–30 amino acids and belong to the superfamily of disulfide-rich conopeptides [88,89][66][67]. In the ω-CnTX family, numerous peptides have been isolated from different conid venoms [90][68]. Depending on the molecular structure, ω-CnTXs that target neuronal N-type CaV channels have been identified as potential drugs for chronic pain treatments. The antagonism with the N-type CaV also suggested a ω-CnTX-neuroprotective effect through a size reduction in cerebral infarction and the delayed inhibition of neuronal cell death in the hippocampal CA1 area [91][69].
PnVIA and PnVIB from Conus pennaceus were among the first ω-CnTx identified to be able to discriminate subtypes of high voltage-activated (HVA) Ca2+ currents in molluscan neurons. In the snail Lymnaea stagnalis, they selectively but reversibly blocked transient HVA currents in caudodorsal cells with negligible effects on L-type currents. Although no clear effects on neurological dysfunctions were reported, they were considered useful selective drugs for relevant CaV channel subtypes [70][48].
2.3. K
V
Channels
KV channels are plasma membrane proteins allowing the selective outside/inside flux of K+ ions in response to the membrane depolarization. KV current activity plays a crucial role in many biological processes and functions such as RP and cell volume regulation, propagation of action potential in nerves, cardiac and skeletal muscles, cell proliferation, differentiation, and apoptosis [93][70]. Furthermore, KV s are essential in the regulation of Ca2+ six transmembrane helices (S1–S6) embedded in the lipid bilayer, forming the voltage sensor domain (S1 to S4) and the pore-forming domain (S5, S6). The voltage sensor domain induces a channel conformational change by sensing RP changes, whereas the interaction with the pore generates K+ ion current fluxes [94][71]. KV channels include 12 different channel families among which a pivotal role is played by the KV1 family, which contains up to eight isoforms (KV1.1–KV1.8). Among them, KV1.3 was first detected in T-cells and, hence, is considered as a possible target for treating autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, and psoriasis. Subsequently, KV1.3 has been proved to be widely distributed in organs and tissues and mainly expressed in both nervous and immune systems participating in several signaling pathways of either normal and/or cancer cells. In particular, KV1.3 channel expressions and/or alterations are involved in numerous pathophysiological processes, such as insulin and apoptosis sensitivity, neoplastic malignancy, inflammatory diseases, cognitive alterations, and anxiety [95][72]. More recently, it has been shown that the inhibitors of the KV1.3 channel reduce neuroinflammation in rodents together with Alzheimer’s and Parkinson’s disease and trauma derived brain injury likely by enabling microglia to resist depolarization stimuli [96][73]. Being involved in overmentioned pathologies, the KV1.3 channel and its blockers have been considered as safe pharmacological tools for chronic inflammatory disease therapies such as type II diabetes mellitus, obesity, and cancer [97][74]. Among the CnTX studied so far, a few can modulate KV channels (Table 3).
Table 3.
CnTX subfamilies targeting voltage-gated potassium (K
V
) channel subtypes, functional impact, and pathophysiological activity.
In 1998, two novel peptides from the venoms of Conus striatus and Conus purpurascens were purified and characterized, demonstrating their ability to bind and block KV channels. kA-CnTX SIVA caused peculiar spastic paralytic symptoms in fish and repetitive action potential oscillations in amphibian nerve-muscle tissues in response to exposure and injection [98][75]. The latter κ-CnTX PVIIA, belonging to a different family, was first described to bind and block K+ channels [99][76]. This peptide possesses a disulfide bridge pattern similar to those of ω- and δ-CnTX [108][85].
In addition, KV1.3 blockers can ameliorate several harmful diseases, such as rheumatoid arthritis, diabetes, and dermatitis in animal models with a safety profile in rodents and primates [100][77]. Accurate molecular simulation techniques aimed to disclose the interaction between PVIIA and shaker KV channels demonstrated the existence of two clusters of amino acids that are critical for the binding between the toxin and the ion channel. The consistency of this binding model and the experimental data indicate that the interaction between PVIIA-KV and shaker KV channels may be useful for the development of new therapeutic agents [109][86].
kM-CnTX RIIIK is a 24 amino acid peptide isolated from Conus radiatus venom identified as the first CnTX able to block human KV1.2 channels. Although structurally similar to µ-CnTX GIIIA, RIIIK inhibits shaker KV expressed in Xenopus oocytes, whereas it showed no affinity with the mammalian KV1.1, KV1.3, and KV1.4 subtypes [101][78]. When administered before reperfusion, RIIIK significantly reduced the in vivo infarct size in rat hearts demonstrating a potential cardioprotective action. On the contrary, another K-CnTX RIIIJ from the same conid venom did not exert any clear cardio-protective effects when targeting KV1.2–KV1.5. However, both isoforms were suggested to provide new hints for understanding the biological mechanism of cardioprotection [102][79].
References
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone Snails: A Big Store of Conotoxins for Novel Drug Discovery. Toxins 2017, 9, 397.
- Layer, R.; McIntosh, J. Conotoxins: Therapeutic Potential and Application. Mar. Drugs 2006, 4, 119–142.
- Terlau, H.; Olivera, B.M. Conus Venoms: A Rich Source of Novel Ion Channel-Targeted Peptides. Physiol. Rev. 2004, 84, 41–68.
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509.
- Robinson, S.; Norton, R. Conotoxin Gene Superfamilies. Mar. Drugs 2014, 12, 6058–6101.
- Halai, R.; Craik, D.J. Conotoxins: Natural product drug leads. Nat. Prod. Rep. 2009, 26, 526.
- Jin, A.-H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549.
- Lewis, R.J. Conotoxins: Molecular and Therapeutic Targets. In Marine Toxins as Research Tools; Fusetani, N., Kem, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 46, pp. 45–65.
- Duggan, P.; Tuck, K. Bioactive Mimetics of Conotoxins and other Venom Peptides. Toxins 2015, 7, 4175–4198.
- Gallo, A.; Boni, R.; Tosti, E. Neurobiological activity of conotoxins via sodium channel modulation. Toxicon 2020, 187, 47–56.
- Tosti, E.; Boni, R.; Gallo, A. µ-Conotoxins Modulating Sodium Currents in Pain Perception and Transmission: A Therapeutic Potential. Mar. Drugs 2017, 15, 295.
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154.
- Mackieh, R.; Abou-Nader, R.; Wehbe, R.; Mattei, C.; Legros, C.; Fajloun, Z.; Sabatier, J.M. Voltage-Gated Sodium Channels: A Prominent Target of Marine Toxins. Mar. Drugs 2021, 19, 562.
- Catterall, W.A. From Ionic Currents to Molecular Mechanisms. Neuron 2000, 26, 13–25.
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 2005, 57, 397–409.
- Mattei, C.; Legros, C. The voltage-gated sodium channel: A major target of marine neurotoxins. Toxicon 2014, 91, 84–95.
- O’Malley, H.A.; Isom, L.L. Sodium Channel β Subunits: Emerging Targets in Channelopathies. Annu. Rev. Physiol. 2015, 77, 481–504.
- Deuis, J.R.; Mueller, A.; Israel, M.R.; Vetter, I. The pharmacology of voltage-gated sodium channel activators. Neuropharmacology 2017, 127, 87–108.
- Cummins, T.R.; Aglieco, F.; Dib-Hajj, S.D. Critical Molecular Determinants of Voltage-Gated Sodium Channel Sensitivity to μ-Conotoxins GIIIA/B. Mol. Pharmacol. 2002, 61, 1192–1201.
- Green, B.R.; Olivera, B.M. Venom Peptides From Cone Snails. In Current Topics in Membranes; Elsevier: Amsterdam, The Netherlands, 2016; Volume 78, pp. 65–86.
- Favreau, P.; Benoit, E.; Hocking, H.G.; Carlier, L.; D’hoedt, D.; Leipold, E.; Markgraf, R.; Schlumberger, S.; Córdova, M.A.; Gaertner, H.; et al. A novel µ-conopeptide, CnIIIC, exerts potent and preferential inhibition of NaV1.2/1.4 channels and blocks neuronal nicotinic acetylcholine receptors: A µ-conopeptide with novel neuropharmacology. Br. J. Pharmacol. 2012, 166, 1654–1668.
- Zhang, M.-M.; Fiedler, B.; Green, B.R.; Catlin, P.; Watkins, M.; Garrett, J.E.; Smith, B.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Structural and Functional Diversities among μ-Conotoxins Targeting TTX-resistant Sodium Channels. Biochemistry 2006, 45, 3723–3732.
- Safo, P.; Rosenbaum, T.; Shcherbatko, A.; Choi, D.-Y.; Han, E.; Toledo-Aral, J.J.; Olivera, B.M.; Brehm, P.; Mandel, G. Distinction among Neuronal Subtypes of Voltage-Activated Sodium Channels by μ-Conotoxin PIIIA. J. Neurosci. 2000, 20, 76–80.
- Wang, C.-Z.; Zhang, H.; Jiang, H.; Lu, W.; Zhao, Z.-Q.; Chi, C.-W. A novel conotoxin from Conus striatus, μ-SIIIA, selectively blocking rat tetrodotoxin-resistant sodium channels. Toxicon 2006, 47, 122–132.
- West, P.J.; Bulaj, G.; Garrett, J.E.; Olivera, B.M.; Yoshikami, D. μ-Conotoxin SmIIIA, a Potent Inhibitor of Tetrodotoxin-Resistant Sodium Channels in Amphibian Sympathetic and Sensory Neurons. Biochemistry 2002, 41, 15388–15393.
- Yao, S.; Zhang, M.-M.; Yoshikami, D.; Azam, L.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure, Dynamics, and Selectivity of the Sodium Channel Blocker μ-Conotoxin SIIIA. Biochemistry 2008, 47, 10940–10949.
- Khoo, K.K.; Feng, Z.-P.; Smith, B.J.; Zhang, M.-M.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure of the Analgesic μ-Conotoxin KIIIA and Effects on the Structure and Function of Disulfide Deletion. Biochemistry 2009, 48, 1210–1219.
- Schroeder, C.I.; Adams, D.; Thomas, L.; Alewood, P.F.; Lewis, R.J. N- and c-terminal extensions of μ-conotoxins increase potency and selectivity for neuronal sodium channels. Biopolymers 2012, 98, 161–165.
- Zhang, M.-M.; Green, B.R.; Catlin, P.; Fiedler, B.; Azam, L.; Chadwick, A.; Terlau, H.; McArthur, J.R.; French, R.J.; Gulyas, J.; et al. Structure/Function Characterization of μ-Conotoxin KIIIA, an Analgesic, Nearly Irreversible Blocker of Mammalian Neuronal Sodium Channels. J. Biol. Chem. 2007, 282, 30699–30706.
- Knapp, O.; McArthur, J.R.; Adams, D.J. Conotoxins Targeting Neuronal Voltage-Gated Sodium Channel Subtypes: Potential Analgesics? Toxins 2012, 4, 1236–1260.
- Fiedler, B.; Zhang, M.-M.; Buczek, O.; Azam, L.; Bulaj, G.; Norton, R.S.; Olivera, B.M.; Yoshikami, D. Specificity, affinity and efficacy of iota-conotoxin RXIA, an agonist of voltage-gated sodium channels NaV 1.2, 1.6 and 1.7. Biochem. Pharmacol. 2008, 75, 2334–2344.
- Shen, H.; Li, Z.; Jiang, Y.; Pan, X.; Wu, J.; Cristofori-Armstrong, B.; Smith, J.J.; Chin, Y.K.Y.; Lei, J.; Zhou, Q.; et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362, eaau2596.
- Tikhonov, D.B.; Zhorov, B.S. Predicting Structural Details of the Sodium Channel Pore Basing on Animal Toxin Studies. Front. Pharmacol. 2018, 9, 880.
- Hui, K.; Lipkind, G.; Fozzard, H.A.; French, R.J. Electrostatic and Steric Contributions to Block of the Skeletal Muscle Sodium Channel by μ-Conotoxin. J. Gen. Physiol. 2002, 119, 45–54.
- Leipold, E.; DeBie, H.; Zorn, S.; Adolfo, B.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. µO-Conotoxins Inhibit NaV Channels by Interfering with their Voltage Sensors in Domain-2. Channels 2007, 1, 253–262.
- Leipold, E.; Hansel, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. Molecular interaction of δ-conotoxins with voltage-gated sodium channels. FEBS Lett. 2005, 579, 3881–3884.
- Green, B.R.; Bulaj, G.; Norton, R.S. Structure and function of μ-conotoxins, peptide-based sodium channel blockers with analgesic activity. Future Med. Chem. 2014, 6, 1677–1698.
- Li, R.A.; Tomaselli, G.F. Using the deadly μ-conotoxins as probes of voltage-gated sodium channels. Toxicon 2004, 44, 117–122.
- Finol-Urdaneta, R.K.; McArthur, J.R.; Korkosh, V.S.; Huang, S.; McMaster, D.; Glavica, R.; Tikhonov, D.B.; Zhorov, B.S.; French, R.J. Extremely Potent Block of Bacterial Voltage-Gated Sodium Channels by µ-Conotoxin PIIIA. Mar. Drugs 2019, 17, 510.
- Berridge, M.J. Neuronal Calcium Signaling. Neuron 1998, 21, 13–26.
- Zamponi, G.W.; Lewis, R.J.; Todorovic, S.M.; Arneric, S.P.; Snutch, T.P. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res. Rev. 2009, 60, 84–89.
- Mochida, S. Presynaptic Calcium Channels. Int. J. Mol. Sci. 2019, 20, 2217.
- Tosti, E. Calcium ion currents mediating oocyte maturation events. Reprod. Biol. Endocrinol. 2006, 4, 26.
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947.
- Bourinet, E.; Zamponi, G.W. Block of voltage-gated calcium channels by peptide toxins. Neuropharmacology 2017, 127, 109–115.
- Dolphin, A.C. Voltage-gated calcium channels and their auxiliary subunits: Physiology and pathophysiology and pharmacology: Voltage-gated calcium channels. J. Pharmacol. 2016, 594, 5369–5390.
- Vink, S.; Alewood, P. Targeting voltage-gated calcium channels: Developments in peptide and small-molecule inhibitors for the treatment of neuropathic pain: VGCC ligands and pain. Br. J. Pharmacol. 2012, 167, 970–989.
- Kits, K.S.; Lodder, J.C.; Van Der Schors, R.C.; Li, K.W.; Geraerts, W.P.; Fainzilber, M. Novel ω-Conotoxins Block Dihydropyridine—Insensitive High Voltage—Activated Calcium Channels in Molluscan Neurons. J. Neurochem. 1996, 67, 2155–2163.
- Fainzilber, M.; Lodder, J.C.; van der Schors, R.C.; Li, K.W.; Yu, Z.; Burlingame, A.L.; Geraerts, W.P.M.; Kits, K.S. A Novel Hydrophobic ω-Conotoxin Blocks Molluscan Dihydropyridine-Sensitive Calcium Channels. Biochemistry 1996, 35, 8748–8752.
- Kerr, L.M.; Yoshikami, D. A venom peptide with a novel presynaptic blocking action. Nature 1984, 308, 282–284.
- Patel, R.; Montagut-Bordas, C.; Dickenson, A.H. Calcium channel modulation as a target in chronic pain control: Calcium channel antagonists and chronic pain. Br. J. Pharmacol. 2018, 175, 2173–2184.
- Oliveira, K.M.; Lavor, M.S.L.; Silva, C.M.O.; Fukushima, F.B.; Rosado, I.R.; Silva, J.F.; Martins, B.C.; Guimarães, L.B.; Gomez, M.V.; Melo, M.M. Omega-conotoxin MVIIC attenuates neuronal apoptosis in vitro and improves significant recovery after spinal cord injury in vivo in rats. Int. J. Clin. Exp. 2014, 7, 3524.
- Sousa, S.R.; McArthur, J.R.; Brust, A.; Bhola, R.F.; Rosengren, K.J.; Ragnarsson, L.; Dutertre, S.; Alewood, P.F.; Christie, M.J.; Adams, D.J.; et al. Novel analgesic ω-conotoxins from the vermivorous cone snail Conus moncuri provide new insights into the evolution of conopeptides. Sci. Rep. 2018, 8, 13397.
- Ramilo, C.A.; Zafaralla, G.C.; Nadasdi, L.; Hammerland, L.G.; Yoshikami, D.; Gray, W.R.; Kristipati, R.; Ramachandran, J.; Miljanich, G. Novel α- and ω-conotoxins and Conus striatus venom. Biochemistry 1992, 31, 9919–9926.
- Wang, F.; Yan, Z.; Liu, Z.; Wang, S.; Wu, Q.; Yu, S.; Ding, J.; Dai, Q. Molecular basis of toxicity of N-type calcium channel inhibitor MVIIA. Neuropharmacology 2016, 101, 137–145.
- Berecki, G.; Motin, L.; Haythornthwaite, A.; Vink, S.; Bansal, P.; Drinkwater, R.; Wang, C.I.; Moretta, M.; Lewis, R.J.; Alewood, P.F.; et al. Analgesic ω-Conotoxins CVIE and CVIF Selectively and Voltage-Dependently Block Recombinant and Native N-Type Calcium Channels. Mol. Pharmacol. 2010, 77, 139–148.
- Lewis, R.J.; Nielsen, K.J.; Craik, D.J.; Loughnan, M.L.; Adams, D.A.; Sharpe, I.A.; Luchian, T.; Adams, D.J.; Bond, T.; Thomas, L.; et al. Novel ω-Conotoxins from Conus catus Discriminate among Neuronal Calcium Channel Subtypes. Int. J. Biol. Chem. 2000, 275, 35335–35344.
- Schroeder, C.; Doering, C.; Zamponi, G.; Lewis, R. N-type Calcium Channel Blockers: Novel Therapeutics for the Treatment of Pain. J. Med. Chem. 2006, 2, 535–543.
- Lee, S.; Kim, Y.; Back, S.K.; Choi, H.-W.; Lee, J.Y.; Jung, H.H.; Ryu, J.H.; Suh, H.-W.; Na, H.S.; Kim, H.J.; et al. Analgesic Effect of Highly Reversible ω-Conotoxin FVIA on N Type Ca2+ Channels. Mol. Pain 2010, 6, 1744–8069.
- Favreau, P.; Gilles, N.; Lamthanh, H.; Bournaud, R.; Shimahara, T.; Bouet, F.; Laboute, P.; Letourneux, Y.; Ménez, A.; Molgó, J.; et al. A New ω-Conotoxin That Targets N-Type Voltage-Sensitive Calcium Channels with Unusual Specificity. Biochemistry 2001, 40, 14567–14575.
- Daly, N.L.; Callaghan, B.; Clark, R.J.; Nevin, S.T.; Adams, D.J.; Craik, D.J. Structure and Activity of α-Conotoxin PeIA at Nicotinic Acetylcholine Receptor Subtypes and GABAB Receptor-coupled N-type Calcium Channels. J. Biol. Chem. 2011, 286, 10233–10237.
- Cai, F.; Xu, N.; Liu, Z.; Ding, R.; Yu, S.; Dong, M.; Wang, S.; Shen, J.; Tae, H.-S.; Adams, D.J.; et al. Targeting of N-Type Calcium Channels via GABAB-Receptor Activation by α-Conotoxin Vc1.1 Variants Displaying Improved Analgesic Activity. J. Med. Chem. 2018, 61, 10198–10205.
- Cuny, H.; de Faoite, A.; Huynh, T.G.; Yasuda, T.; Berecki, G.; Adams, D.J. γ-Aminobutyric Acid Type B (GABAB) Receptor Expression Is Needed for Inhibition of N-type (CaV2.2) Calcium Channels by Analgesic α-Conotoxins. J. Biol. Chem. 2012, 287, 23948–23957.
- Klimis, H.; Adams, D.J.; Callaghan, B.; Nevin, S.; Alewood, P.F.; Vaughan, C.W.; Mozar, C.A.; Christie, M.J. A novel mechanism of inhibition of high-voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 2011, 152, 259–266.
- Sadeghi, M.; McArthur, J.R.; Finol-Urdaneta, R.K.; Adams, D.J. Analgesic conopeptides targeting G protein-coupled receptors reduce excitability of sensory neurons. Neuropharmacology 2017, 127, 116–123.
- Adams, D.J.; Callaghan, B.; Berecki, G. Analgesic conotoxins: Block and G protein-coupled receptor modulation of N-type (CaV2.2) calcium channels: Conotoxin modulation of calcium channel function. Br. J. Pharmacol. 2012, 166, 486–500.
- Hannon, H.; Atchison, W. Omega-Conotoxins as Experimental Tools and Therapeutics in Pain Management. Mar. Drugs 2013, 11, 680–699.
- Ramírez, D.; Gonzalez, W.; Fissore, R.; Carvacho, I. Conotoxins as Tools to Understand the Physiological Function of Voltage-Gated Calcium (CaV) Channels. Mar. Drugs 2017, 15, 313.
- Ito, Y.; Araki, N. Calcium antagonists: Current and future applications based on new evidence. Neuroprotective effect of calcium antagonists. Clin. Calcium 2010, 20, 83–88.
- Carvalho-de-Souza, J.L.; Saponaro, A.; Bassetto, C.A.Z.; Rauh, O.; Schroeder, I.; Franciolini, F.; Catacuzzeno, L.; Bezanilla, F.; Thiel, G.; Moroni, A. Experimental challenges in ion channel research: Uncovering basic principles of permeation and gating in potassium channels. Adv. Phys. X 2022, 7, 1978317.
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693.
- Serrano-Albarrás, A.; Estadella, I.; Cirera-Rocosa, S.; Navarro-Pérez, M.; Felipe, A. KV1.3: A multifunctional channel with many pathological implications. Expert Opin. Ther. Targets 2018, 22, 101–105.
- Fomina, A.F.; Nguyen, H.M.; Wulff, H. KV1.3 inhibition attenuates neuroinflammation through disruption of microglial calcium signaling. Channels 2021, 15, 67–78.
- Pérez-Verdaguer, M.; Capera, J.; Serrano-Novillo, C.; Estadella, I.; Sastre, D.; Felipe, A. The voltage-gated potassium channel KV1.3 is a promising multitherapeutic target against human pathologies. Expert Opin. Ther. Targets 2016, 20, 577–591.
- Craig, A.G.; Zafaralla, G.; Cruz, L.J.; Santos, A.D.; Hillyard, D.R.; Dykert, J.; Rivier, J.E.; Gray, W.R.; Imperial, J.; DelaCruz, R.G.; et al. An O-Glycosylated Neuroexcitatory Conus Peptide. Biochemistry 1998, 37, 16019–16025.
- Shon, K.-J.; Stocker, M.; Terlau, H.; Stühmer, W.; Jacobsen, R.; Walker, C.; Grilley, M.; Watkins, M.; Hillyard, D.R.; Gray, W.R.; et al. κ-Conotoxin Pviia Is a Peptide Inhibiting theShaker K+ Channel. Int. J. Biol. Chem. 1998, 273, 33–38.
- Rangaraju, S.; Chi, V.; Pennington, M.W.; Chandy, K.G. KV1.3 potassium channels as a therapeutic target in multiple sclerosis. Expert Opin. Ther. Targets 2009, 13, 909–924.
- Ferber, M.; Al-Sabi, A.; Stocker, M.; Olivera, B.M.; Terlau, H. Identification of a mammalian target of κM-conotoxin RIIIK. Toxicon 2004, 43, 915–921.
- Chen, P.; Dendorfer, A.; Finol-Urdaneta, R.K.; Terlau, H.; Olivera, B.M. Biochemical Characterization of κM-RIIIJ, a KV1.2 Channel Blocker. J. Biol. Chem. 2010, 285, 14882–14889.
- Finol-Urdaneta, R.K.; Belovanovic, A.; Micic-Vicovac, M.; Kinsella, G.K.; McArthur, J.R.; Al-Sabi, A. Marine Toxins Targeting KV1 Channels: Pharmacological Tools and Therapeutic Scaffolds. Mar. Drugs 2020, 18, 173.
- Kauferstein, S.; Huys, I.; Kuch, U.; Melaun, C.; Tytgat, J.; Mebs, D. Novel conopeptides of the I-superfamily occur in several clades of cone snails. Toxicon 2004, 44, 539–548.
- Liu, Z.; Xu, N.; Hu, J.; Zhao, C.; Yu, Z.; Dai, Q. Identification of novel I-superfamily conopeptides from several clades of Conus species found in the South China Sea. Peptides 2009, 30, 1782–1787.
- Kauferstein, S.; Huys, I.; Lamthanh, H.; Stöcklin, R.; Sotto, F.; Menez, A.; Tytgat, J.; Mebs, D. A novel conotoxin inhibiting vertebrate voltage-sensitive potassium channels. Toxicon 2003, 42, 43–52.
- Lubbers, N.L.; Campbell, T.J.; Polakowski, J.S.; Bulaj, G.; Layer, R.T.; Moore, J.; Gross, G.J.; Cox, B.F. Postischemic Administration of CGX-1051, a Peptide from Cone Snail Venom, Reduces Infarct Size in Both Rat and Dog Models of Myocardial Ischemia and Reperfusion. J. Cardiovasc. Pharmacol. 2005, 46, 141–146.
- Savarin, P.; Guenneugues, M.; Gilquin, B.; Lamthanh, H.; Gasparini, S.; Zinn-Justin, S.; Ménez, A. Three-Dimensional Structure of κ-Conotoxin PVIIA, a Novel Potassium Channel-Blocking Toxin from Cone Snails. Biochemistry 1998, 37, 5407–5416.
- Moran, O. Molecular simulation of the interaction of κ-conotoxin-PVIIA with the Shaker potassium channel pore. Eur. Biophys. 2001, 30, 528–536.
More