Cholestatic diseases can be caused by the dysfunction of transporters involved in hepatobiliary circulation. Although pharmacological treatments constitute the current standard of care for these diseases, none are curative, with liver transplantation being the only long-term solution for severe cholestasis, albeit with many disadvantages. Liver-directed gene therapy has shown promising results in clinical trials for genetic diseases, and it could constitute a potential new therapeutic approach for cholestatic diseases. Gene therapy has emerged as a promising approach to achieve safe, stable, and efficient long-term correction for a wide range of genetic diseases, including monogenic liver disorders, for which liver transplantation remains the only cure, as well as acquired liver diseases.
- cholestatic diseases
- gene therapy
- AAV
- PFIC
1. Gene Therapy for Acquired Cholestasis
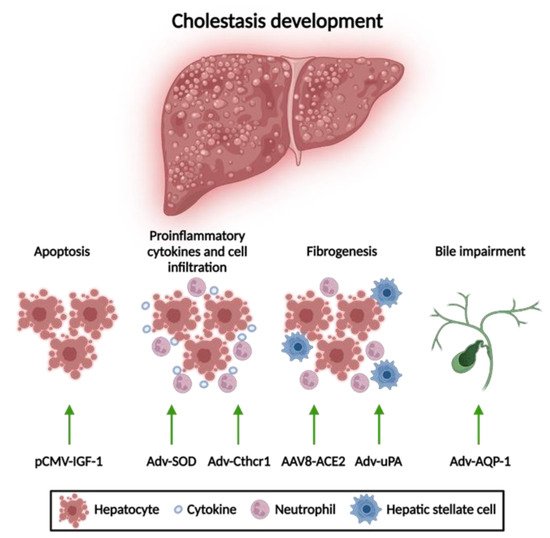
1.1. Apoptosis Attenuation
1.2. Reduction of Mitochondrial Oxidative Stress
1.3. Anti-Fibrotic Therapies
1.4. Amelioration of Bile Flow
2. Gene Therapy for Inherited Cholestasis
2.1. Gene Therapy of Genetic Disorders with Associated Cholestasis
2.2. Gene Therapy for PFIC Diseases
Gene Therapy for PFIC3 Based on ABCB4 Supplementation
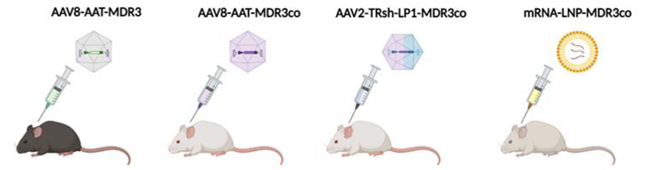
| Aronson et al. [22] | Weber et al. [26] | Siew et al. [25] | Wei et al. [23] | |
|---|---|---|---|---|
| Strain Background | C57BL/6 Abcb4-/- | FVB Abcb4-/- | FVB Abcb4-/- | BALB/c Abcb4-/- |
| Phenotype | Mild (requiring cholate-enriched diet) |
Severe (similar to patients) |
Severe (similar to patients) |
More severe |
| Vector | AAV8 | AAV8 | Hybrid AAV-piggyBac transposon | LNP |
| Dose | 5 × 1013 vg/kg | 1 × 1014 vg/kg | ~2 × 1014 vg/kg | 1.0 mg/kg |
| Age of treatment | 10-week-old | 2- or 5-week-old | Newborn | 4-week-old |
| Outcomes | Increased biliary PC and cholesterol content. Rescue of serum ALT, ALP and bilirubin levels. Prevention of liver fibrosis. | Increased biliary PC. Rescue of serum transaminases, ALP and BA levels. Improvement of the degree of hepatosplenomegaly. Prevention and reversal of liver fibrosis. | Increased biliary PC. Decreased hepatomegaly and serum parameters (ALT, ALP, BAs). Reduced liver fibrosis and liver tumor incidence. | Increased biliary PC (10–25% WT) and %BW. Decreased hepatomegaly and serum parameters (ALT, ALP, BAs). Normalization of liver fibrosis and portal hypertension. |
| Advantages | Long-term correction. No risk of mutagenesis. | Granted orphan drug designation. Long-term prevention and correction at early and late stages of PFIC3, respectively. No risk of mutagenesis. | Long-term correction. Preventing genome loss by hepatocellular proliferation during liver growth. | No risk of mutagenesis. Less immune responses. |
| Disadvantages | Need for challenge with BA-enriched dietary supplementation (model). Need to evaluate efficacy in younger mice more representative of the age of patients. Risks of using a high viral dose. | Loss of long-term therapeutic effect in half of the females treated with a single dose. Need to address the immune response based on anti-AAV neutralizing antibody for repeated administrations of the vector. Risks of using a high viral dose. | Risk of mutagenesis. Transposase overexpression Lack of serotype that efficiently transduces human hepatocytes. |
Less durable expression. Requires repeated parenteral dosing. |
| Aronson et al. [112] | Weber et al. [113] | Siew et al. [116] | Wei et al. [114] | |
|---|---|---|---|---|
| Strain Background | C57BL/6 Abcb4-/- | FVB Abcb4-/- | FVB Abcb4-/- | BALB/c Abcb4-/- |
| Phenotype | Mild (requiring cholate-enriched diet) |
Severe (similar to patients) |
Severe (similar to patients) |
More severe |
| Vector | AAV8 | AAV8 | Hybrid AAV-piggyBac transposon | LNP |
| Dose | 5 × 1013 vg/kg | 1 × 1014 vg/kg | ~2 × 1014 vg/kg | 1.0 mg/kg |
| Age of treatment | 10-week-old | 2- or 5-week-old | Newborn | 4-week-old |
| Outcomes | Increased biliary PC and cholesterol content. Rescue of serum ALT, ALP and bilirubin levels. Prevention of liver fibrosis. | Increased biliary PC. Rescue of serum transaminases, ALP and BA levels. Improvement of the degree of hepatosplenomegaly. Prevention and reversal of liver fibrosis. | Increased biliary PC. Decreased hepatomegaly and serum parameters (ALT, ALP, BAs). Reduced liver fibrosis and liver tumor incidence. | Increased biliary PC (10–25% WT) and %BW. Decreased hepatomegaly and serum parameters (ALT, ALP, BAs). Normalization of liver fibrosis and portal hypertension. |
| Advantages | Long-term correction. No risk of mutagenesis. | Granted orphan drug designation. Long-term prevention and correction at early and late stages of PFIC3, respectively. No risk of mutagenesis. | Long-term correction. Preventing genome loss by hepatocellular proliferation during liver growth. | No risk of mutagenesis. Less immune responses. |
| Disadvantages | Need for challenge with BA-enriched dietary supplementation (model). Need to evaluate efficacy in younger mice more representative of the age of patients. Risks of using a high viral dose. | Loss of long-term therapeutic effect in half of the females treated with a single dose. Need to address the immune response based on anti-AAV neutralizing antibody for repeated administrations of the vector. Risks of using a high viral dose. | Risk of mutagenesis. Transposase overexpression Lack of serotype that efficiently transduces human hepatocytes. |
Less durable expression. Requires repeated parenteral dosing. |
Gene Therapy for PFIC3 Targeting Mechanisms of Disease
Gene Therapy for Other Types of PFIC
References
- Mariotti, V.; Strazzabosco, M.; Fabris, L.; Calvisi, D.F. Animal Models of Biliary Injury and Altered Bile Acid Metabolism. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 1254–1261.
- Fernández-Ramos, D.; Fernández-Tussy, P.; Lopitz-Otsoa, F.; Gutiérrez-de-Juan, V.; Navasa, N.; Barbier-Torres, L.; Zubiete-Franco, I.; Simón, J.; Fernández, A.F.; Arbelaiz, A.; et al. MiR-873-5p Acts as an Epigenetic Regulator in Early Stages of Liver Fibrosis and Cirrhosis. Cell Death Dis. 2018, 9, 958.
- Wang, K.P.-C.; Lee, L.-M.; Lin, T.-J.; Sheen-Chen, S.-M.; Lin, J.-W.; Chiu, W.-T.; Wang, C.-C.; Hung, K.-S. Gene Transfer of IGF1 Attenuates Hepatocellular Apoptosis After Bile Duct Ligation. J. Surg. Res. 2011, 167, 237–244.
- Zhong, Z.; Froh, M.; Wheeler, M.; Smutney, O.; Lehmann, T.; Thurman, R. Viral Gene Delivery of Superoxide Dismutase Attenuates Experimental Cholestasis-Induced Liver Fibrosis in the Rat. Gene Ther. 2002, 9, 183–191.
- Bian, Z.; Miao, Q.; Zhong, W.; Zhang, H.; Wang, Q.; Peng, Y.; Chen, X.; Guo, C.; Shen, L.; Yang, F.; et al. Treatment of Cholestatic Fibrosis by Altering Gene Expression of Cthrc1: Implications for Autoimmune and Non-Autoimmune Liver Disease. J. Autoimmun. 2015, 63, 76–87.
- Miranda-Díaz, A.; Rincón, A.R.; Salgado, S.; Vera-Cruz, J.; Gálvez, J.; Islas, M.C.; Berumen, J.; Aguilar Cordova, E.; Armendáriz-Borunda, J. Improved Effects of Viral Gene Delivery of Human UPA plus Biliodigestive Anastomosis Induce Recovery from Experimental Biliary Cirrhosis. Mol. Ther. 2004, 9, 30–37.
- Salgado, S.; Garcia, J.; Vera, J.; Siller, F.; Bueno, M.; Miranda, A.; Segura, A.; Grijalva, G.; Segura, J.; Orozco, H.; et al. Liver Cirrhosis Is Reverted by Urokinase-Type Plasminogen Activator Gene Therapy. Mol. Ther. 2000, 2, 545–551.
- Mak, K.Y.; Chin, R.; Cunningham, S.C.; Habib, M.R.; Torresi, J.; Sharland, A.F.; Alexander, I.E.; Angus, P.W.; Herath, C.B. ACE2 Therapy Using Adeno-Associated Viral Vector Inhibits Liver Fibrosis in Mice. Mol. Ther. 2015, 23, 1434–1443.
- Yang, T.; Poenisch, M.; Khanal, R.; Hu, Q.; Dai, Z.; Li, R.; Song, G.; Yuan, Q.; Yao, Q.; Shen, X.; et al. Therapeutic HNF4A MRNA Attenuates Liver Fibrosis in a Preclinical Model. J. Hepatol. 2021, 75, 1420–1433.
- Marrone, J.; Lehmann, G.L.; Soria, L.R.; Pellegrino, J.M.; Molinas, S.; Marinelli, R.A. Adenoviral Transfer of Human Aquaporin -1 Gene to Rat Liver Improves Bile Flow in Estrogen-Induced Cholestasis. Gene Ther. 2014, 21, 1058–1064.
- Marrone, J.; Soria, L.R.; Danielli, M.; Lehmann, G.L.; Larocca, M.C.; Marinelli, R.A. Hepatic Gene Transfer of Human Aquaporin-1 Improves Bile Salt Secretory Failure in Rats with Estrogen-induced Cholestasis. Hepatology 2016, 64, 535–548.
- Nathwani, A.C.; Tuddenham, E.G.D.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365.
- Maestro, S.; Weber, N.D.; Zabaleta, N.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Novel Vectors and Approaches for Gene Therapy in Liver Diseases. JHEP Rep. 2021, 3, 100300.
- Lumbreras, S.; Ricobaraza, A.; Baila-Rueda, L.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Uriarte, I.; Bunuales, M.; Avila, M.A.; Monte, M.J.; Marin, J.J.G.; et al. Gene Supplementation of CYP27A1 in the Liver Restores Bile Acid Metabolism in a Mouse Model of Cerebrotendinous Xanthomatosis. Mol. Ther.-Methods Clin. Dev. 2021, 22, 210–221.
- Collaud, F.; Bortolussi, G.; Guianvarc’h, L.; Aronson, S.J.; Bordet, T.; Veron, P.; Charles, S.; Vidal, P.; Sola, M.S.; Rundwasser, S.; et al. Preclinical Development of an AAV8-HUGT1A1 Vector for the Treatment of Crigler-Najjar Syndrome. Mol. Ther.-Methods Clin. Dev. 2019, 12, 157–174.
- Ronzitti, G.; Bortolussi, G.; van Dijk, R.; Collaud, F.; Charles, S.; Leborgne, C.; Vidal, P.; Martin, S.; Gjata, B.; Sola, M.S.; et al. A Translationally Optimized AAV-UGT1A1 Vector Drives Safe and Long-Lasting Correction of Crigler-Najjar Syndrome. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16049.
- Ginocchio, V.M.; Ferla, R.; Auricchio, A.; Brunetti-Pierri, N. Current Status on Clinical Development of Adeno-Associated Virus-Mediated Liver-Directed Gene Therapy for Inborn Errors of Metabolism. Hum. Gene Ther. 2019, 30, 1204–1210.
- Bull, L.N.; Thompson, R.J. Progressive Familial Intrahepatic Cholestasis. Clin. Liver Dis. 2018, 22, 657–669.
- Bosma, P.J.; Wits, M.; Oude-Elferink, R.P.J. Gene Therapy for Progressive Familial Intrahepatic Cholestasis: Current Progress and Future Prospects. Int. J. Mol. Sci. 2020, 22, 273.
- de Vree, J.M.L.; Ottenhoff, R.; Bosma, P.J.; Smith, A.J.; Aten, J.; Oude Elferink, R.P.J. Correction of Liver Disease by Hepatocyte Transplantation in a Mouse Model of Progressive Familial Intrahepatic Cholestasis. Gastroenterology 2000, 119, 1720–1730.
- Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Yoshida, S.; Nasser, I.; Ke, Q.; Kang, P.M.; Popov, Y. A New Mdr2−/− Mouse Model of Sclerosing Cholangitis with Rapid Fibrosis Progression, Early-Onset Portal Hypertension, and Liver Cancer. Am. J. Pathol. 2015, 185, 325–334.
- Aronson, S.J.; Bakker, R.S.; Shi, X.; Duijst, S.; Bloemendaal, L.T.; de Waart, D.R.; Verheij, J.; Ronzitti, G.; Elferink, R.P.O.; Beuers, U.; et al. Liver-Directed Gene Therapy Results in Long-Term Correction of Progressive Familial Intrahepatic Cholestasis Type 3 in Mice. J. Hepatol. 2019, 71, 153–162.
- Wei, G.; Cao, J.; Huang, P.; An, P.; Badlani, D.; Vaid, K.A.; Zhao, S.; Wang, D.Q.-H.; Zhuo, J.; Yin, L.; et al. Synthetic Human ABCB4 MRNA Therapy Rescues Severe Liver Disease Phenotype in a BALB/c.Abcb4 Mouse Model of PFIC3. J. Hepatol. 2021, 74, 1416–1428.
- Weber, N.D.; Martínez-García, J.; González-Aseguinolaza, G. Comment on “Synthetic Human ABCB4 MRNA Therapy Rescues Severe Liver Disease Phenotype in a BALB/c.Abcb4 Mouse Model of PFIC3”. J. Hepatol. 2022, 76, 749–751.
- Siew, S.M.; Cunningham, S.C.; Zhu, E.; Tay, S.S.; Venuti, E.; Bolitho, C.; Alexander, I.E. Prevention of Cholestatic Liver Disease and Reduced Tumorigenicity in a Murine Model of PFIC Type 3 Using Hybrid AAV-PiggyBac Gene Therapy. Hepatology 2019, 70, 2047–2061.
- Weber, N.D.; Odriozola, L.; Martínez-García, J.; Ferrer, V.; Douar, A.; Bénichou, B.; González-Aseguinolaza, G.; Smerdou, C. Gene Therapy for Progressive Familial Intrahepatic Cholestasis Type 3 in a Clinically Relevant Mouse Model. Nat. Commun. 2019, 10, 5694.
- Rajapaksha, I.G.; Angus, P.W.; Herath, C.B. Current Therapies and Novel Approaches for Biliary Diseases. World J. Gastrointest. Pathophysiol. 2019, 10, 1–10.
- Li, J.; Zhu, X.; Zhang, M.; Zhang, Y.; Ye, S.; Leng, Y.; Yang, T.; Kong, L.; Zhang, H. Limb Expression 1-like (LIX1L) Protein Promotes Cholestatic Liver Injury by Regulating Bile Acid Metabolism. J. Hepatol. 2021, 75, 400–413.
- Ceci, L.; Francis, H.; Zhou, T.; Giang, T.; Yang, Z.; Meng, F.; Wu, N.; Kennedy, L.; Kyritsi, K.; Meadows, V.; et al. Knockout of the Tachykinin Receptor 1 in the Mdr2−/− (Abcb4−/−) Mouse Model of Primary Sclerosing Cholangitis Reduces Biliary Damage and Liver Fibrosis. Am. J. Pathol. 2020, 190, 2251–2266.
- Giang, S.; Gordon, R.L.; Haas, K.B. A Diagnostic Quagmire: PFIC5 Presenting as a Rare Cause of Neonatal Cholestasis. ACG Case Rep. J. 2021, 8, e00558.
- Sambrotta, M.; Strautnieks, S.; Papouli, E.; Rushton, P.; Clark, B.E.; Parry, D.A.; Logan, C.V.; Newbury, L.J.; Kamath, B.M.; Ling, S.; et al. Mutations in TJP2 Cause Progressive Cholestatic Liver Disease. Nat. Genet. 2014, 46, 326–328.
- Overeem, A.W.; Li, Q.; Qiu, Y.; Cartón-García, F.; Leng, C.; Klappe, K.; Dronkers, J.; Hsiao, N.; Wang, J.; Arango, D.; et al. A Molecular Mechanism Underlying Genotype-Specific Intrahepatic Cholestasis Resulting From MYO5B Mutations. Hepatology 2020, 72, 213–229.
- Zhang, Y.; Li, F.; Patterson, A.D.; Wang, Y.; Krausz, K.W.; Neale, G.; Thomas, S.; Nachagari, D.; Vogel, P.; Vore, M.; et al. Abcb11 Deficiency Induces Cholestasis Coupled to Impaired β-Fatty Acid Oxidation in Mice. J. Biol. Chem. 2012, 287, 24784–24794.
- Hu, C.; Busuttil, R.W.; Lipshutz, G.S. RH10 Provides Superior Transgene Expression in Mice When Compared with Natural AAV Serotypes for Neonatal Gene Therapy. J. Gene Med. 2010, 12, 766–778.