Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Roman P. Terekhov and Version 2 by Conner Chen.
Flavonoids are widely occurring secondary metabolites of plants. Currently, there is a trend of article numbers increasing, which focuses on the computer modeling of flavonoid interactions with biological targets. Such studies help to accumulatethe data on lead compounds that can find medicinal implementation, including COVID-19. Flavanonol taxifolin demonstrated wound-healing activity. Luteolin, apigenin, and wogonin, which can be classified as flavones, show induced neutrophil apoptosis and have potential as neutrophil apoptosis-inducing anti-inflammatory, proresolution agents.
- flavonoids
- phytomedicine
- taxifolin
- molecular modeling
- COVID-19
1. Structure—Biological Activity Relationship: Qualitative Analysis
The parent structure of flavonoids is 1,3-diphenylpropane, and the aromatic fragments are designated as ring A and ring B [1][67]. The majority of flavonoid groups are characterized by the heterocycle (ring C) containing oxygen. This ring may be aromatic (flavones, flavonols, etc.) or not (flavanones, flavanonols, etc.). As the rule, carbonyl and several hydroxyl functional groups are present in the molecular structure of flavonoids that can act as a pharmacophores.
The phenolic hydroxyl groups of the studied natural compounds serve as H-bond donors. In cases when the hydrophobic interactions play a key role, the presence of the methoxy group leads to an increase of affinity to the target compared with the hydroxyl group [2][68]. Due to aromatic rings, the π,π-interactions with the side residues of heterocyclic and aromatic α-amino acids (tryptophan, histidine, phenylalanine, and tyrosine) are possible [3][69]. Figure 1 demonstrates all types of interactions.
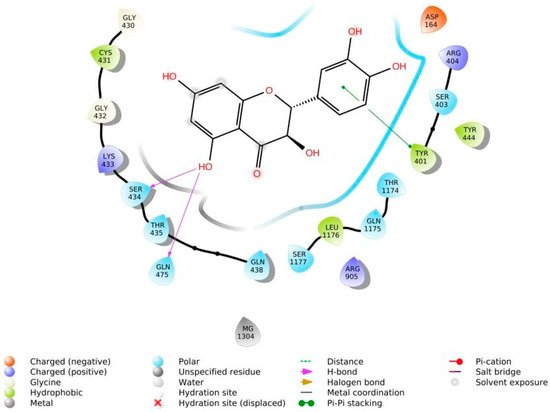
Figure 1.
Interaction of taxifolin and P-glycoprotein.
It was found that the antiangiogenic potential of the flavonoid depends on the presence of a C2-C3 double bond [4][70]; the hydroxyl group in the position 3′ of the ring C contributes to an increase in antioxidant, anti-inflammatory, and antitumor activity [5][71]. If, along with the multiple C2-C3 bonds, a catechol group is present in the ring B, then such a molecule demonstrates a high affinity for the angiotensin-converting enzyme [6][72]. Substituents 3-OH, 5-OH, 6-OMe, 6-OH, 7-OH, 3′-OH, and 4′-OMe were identified as key fragments of the molecules when interacting with multidrug resistance-associated protein 2 (MRP2) [7][55].
It was also interesting to determine the specificity of the interaction of flavonoid groups. Thus, flavones (6-hydroxyluteolin, scutellarein), flavonols (kaempferol), and flavanones (naringenin, eridioctyol) exhibit a high affinity to the estrogen receptor α (ERα), which has been proven in both AutoDock and Glide software. Representatives of these groups of flavonoids can be recommended in the development of antitumor drugs for the treatment of breast cancer [5][8][71,73]. Interaction with this protein target results in several types of patient management, such as estrogen hormone replacement therapy and preventive care for breast cancer [9][74]. Flavones (baicalein, ladanein), flavonols (quercetin), and their glycosylated forms (baicalin) interact with the E protein of various strains of the dengue virus causing fever with a similar name [10][54]. Such ligands may be used in the treatment of this disease [11][75]. It is worth noting that the width of the confidence interval of the scoring function calculated for flavones is quite large. This indicates a different degree of protein-ligand binding within this group. Flavones (5-hydroxyflavone) and flavonols (quercetin) have a high affinity for the potassium channel Kir6.1, acting on which some cardiovascular diseases can be treated [12][56]. Flavones (luteolin, apigenin) can serve as the basis of drugs that control the pathogenicity of Helicobacter pylori due to their ability to bind to one of the main virulence factors of bacteria of this species—vacuolating cytotoxin protein (VacA) [13][76]. Flavonols (quercetin), their glycosides (avicularin, hyperoside), and flavanonols (taxifolin) with comparable effects function as arginase inhibitors, which is a potential target for the development of new approaches to the treatment of leishmaniasis [14][77]. Flavan-3-ols (catechin, epicatechin) are characterized by the best values of the scoring function when binding to the CA II-F complex in comparison with flavones, flavanones, and flavanonols and are of interest for the treatment of fluorosis [15][78]. According to the silico results, flavanones (eriodictyol) and flavanonols (taxifolin) are able to inhibit transcription factors Tec1 and Rfg1 because they can be used in the treatment of infection caused by Candida albicans fungus [16][79].
2. Structure—Biological Activity Relationship: Quantitative Analysis
Meta-analyses of scoring functions calculated during molecular docking was studied in [5][8][10][12][13][14][15][16][54,56,71,73,76,77,78,79]. General information about the average affinities of each flavonoid group to the biological targets is presented in Table 1 and Table 2 for AutoDock and Glide software, respectively.Table 1.
Comparison of the average affinity of flavonoid groups to target proteins in the AutoDock.
| Flavonoid Group | Affinity to the Biological Target, kcal/mol * | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ERα | E Protein DENV2-Thai |
E Protein DENV2-My |
Potassium Channel Kir6.1 | Protein VacA | |||||||
| ERα | Complex CA II-F | Arginase | Tec1 | Rfg1 | |||||||
| Flavones | −8.3 ± 0.6 | −7.8 ± 1.3 | −7.5 ± 0.9 | −6.7 ± n/a | −8.5 ± 0.3 | ||||||
| Flavones | −8.5 ± 0.3 | −3.3 ± 0.0 | - | - | - | ||||||
| Flavonols | −7.9 ± 0.5 | −8.4 ± n/a | |||||||||
| Flavonols | −8.8 ± n/a | - | −8.6 ± n/a | −8.1 ± n/a | - | ||||||
| −8.1 ± n/a | - | - | Flavonol glycosides |
- | −8.1 ± n/a | −7.7 ± n/a | |||||
| Flavonol glycosides | - | - | |||||||||
| - | - | −8.2 ± 0.3 | - | - | Flavanones | −8.5 ± n/a | - | - | - | - | |
| Flavanones | −10.2 ± n/a | −2.7 ± 0.2 | - | −7.7 ± n/a | −6.7 ± n/a | Flavanonols | −9.0 ± n/a | - | - | - | - |
* A lower value of the scoring function corresponds to a better binding.
Table 2.
Comparison of the average affinity of flavonoid groups to target proteins in Glide.
| Flavonoid Group | Affinity to the Biological Target, kcal/mol * | ||||
|---|---|---|---|---|---|
| Flavanonols | |||||
| - | |||||
| −2.9 ± n/a | −8.2 ± n/a | −7.7 ± n/a | −4.9 ± n/a | ||
| Flavan-3-ols | - | −4.7 ± 0.6 | - | - | - |
| Isoflavones | −9.0 ± 0.20 | - | - | - | - |
| Dihydrochalcones | −8.3 ± n/a | - | - | - | - |
* A lower value of the scoring function corresponds to a better binding.
3. Lead Compounds
Molecular docking makes it possible to evaluate the affinity of the ligand with the target. Based on this criterion, it is possible to compile a list of the most active compounds for each specific interaction of a flavonoid with a macromolecule. The structures of the leader compounds are shown in Figure 2. Thus, taxifolin (dihydroquercetin) has a high potential for tuberculosis therapy, as it has demonstrated the ability to interact with DNA gyrase and aminoacyl-tRNA synthetase—two enzymes involved in the translation, transcription, and replication of bacterial DNA [18][97]. The pathogenic effect of the Ebola virus can be disrupted by affecting the vital protein structures VP40, VP35, VP30, and VP24 with flavonoids such as gossypetin or taxifolin. These compounds proved to be leaders in the study of more than 4500 flavonoids [19][98]. It is interesting to notice that despite the near structures of the molecules, they have similar interaction patterns with only two proteins among four targets. Both taxifolin and gossypetin can form H-bonds with His124, Gly126, and Gln170 in VP40. Furthermore, they interact with Gln103, Ser123, Asp124, and Asn185 or Gln184 in the active site of VP24. Key amino acid residues in VP35 are Gln244(2) and Asp302 for taxifolin, while for gossypetin, it is only the Gln241(2). Finally, gossypetin interacts with Asp158(2), Arg196, and Gln233(2) in VP30. However, taxifolin forms H-bonds with Arg196, Gly200(3), Gln233, Ser234, and Phe238 residues of the VP30. Differences in the affinity of these flavonoids are apparently associated with the spatial structure of the heterocyclic ring. Gossypetin has a plane aromatic structure, and the taxifolin molecule is characterized by two chiral centers. Thus, taxifolin can exist as four stereoisomers. Saltillin, taxifolin, and 6-methoxyflavone have a high affinity for N-myristoyltransferase (NMT), a target for the treatment of candidiasis [20][99]. There is also information indicating the feasibility of studying a number of flavonoids in the following diseases: lung cancer [21][100], breast cancer [5][22][50,71], metabolic syndrome [23][49], pathological conditions caused by Pseudomonas aeruginosa [24][101], hypoestrogenism [25][102], and depressive disorders [26][103].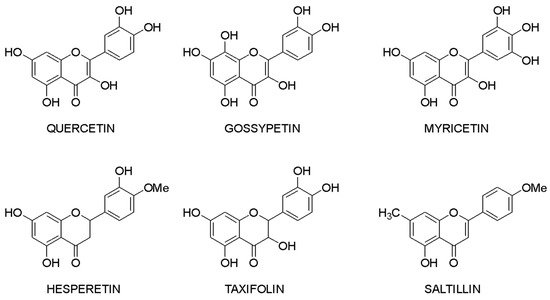
Figure 2.
Lead compounds.