Merkel cell polyomavirus (MCV) is the only known human oncogenic virus in the polyomaviridae family and the etiological agent of most Merkel cell carcinomas (MCC). MCC is an aggressive and highly metastatic skin cancer with a propensity for recurrence and poor prognosis. Large tumor antigen (LT), is an essential oncoprotein for MCV transcription, viral replication, and cancer cell proliferation. MCV LT is a short-lived protein that encodes a unique domain: MCV LT unique regions (MURs). These domains consist of phosphorylation sites that interact with multiple E3 ligases, thus limiting LT expression and consequently, viral replication. In this study, we show that MURs are necessary for regulating LT stability via multiple E3 ligase interactions, resulting in cell growth arrest. While expression of wild-type MCV LT induced a decrease in cellular proliferation, deletion of the MUR domains resulted in increased LT stability and cell proliferation. Conversely, addition of MURs to SV40 LT propagated E3 ligase interactions, which in turn, reduced SV40 LT stability and decreased cell growth activity. Our results demonstrate that compared to other human polyomaviruses (HPyVs), MCV LT has evolved to acquire the MUR domains that are essential for MCV LT autoregulation, potentially leading to viral latency and MCC.
- Merkel cell polyomavirus
- Large T antigen
- Protein stability
1. Introduction
Merkel cell carcinoma (MCC) is a rare but highly aggressive cutaneous neuroendocrine carcinoma [1]. Approximately 80% of Merkel cell carcinomas are caused by Merkel cell polyomavirus (MCV or MCPyV), the only known oncogenic human polyomavirus (HPyV) [2]. MCV is
~5.3 kilobase (kb) viral genome that encodes an early gene locus involved in viral replication, and a late gene locus encoding capsid structure proteins. An early region transcript, MCV large T (LT) protein is generated as a result of alternative splicing of the T antigen locus of the early region [3]. MCV LT is a highly phosphorylated protein that recognizes its viral DNA replication origin and forms a double-hexameric helicase complex to initiate replication of the viral genome [4,5][4][5].
MCV LT is a spliced transcript comprising 2 exons producing a protein 817 amino acids in size [3]. The N-terminal end of MCV LT (1–70 aa) contains the DnaJ domain encoding the specific HPDKGG sequence responsible for Hsc70 binding (42–47 aa) [4] and a conserved LXCXE motif for retinoblastoma (Rb)-binding which has high homology to other polyomaviruses [3]. Similar to other polyomaviruses, MCV LT antigen contains multiple conserved domains including a nuclear localization signal (NLS) [6], the origin-binding domain (OBD) [4], and the carboxyl terminal (C-terminus) half which contains several critical elements essential for DNA binding, helicase activity and viral replication [4,5][4][5]. In contrast to Simian virus 40 (SV40) LT, expression of wild-type MCV LT inhibits cell proliferation [7,8][7][8]. Further analysis of the MCV genome clonally integrated into MCV-positive tumor cells revealed mutations that induce premature truncations in the LT antigen [3], resulting in the deletion of the C-terminus growth-inhibitory domain [7,8][7][8].
Protein phosphorylation is a reversible posttranslational modification essential for regulating protein function [9]. Previous reports have shown that phosphorylation is a regulatory mechanism associated with T antigen regulation in Murine polyomavirus (MPyV) and SV40, known polyomavirus models [10,11,12][10][11][12]. The phosphorylation of SV40 LT and MPyV LT is essential for regulating LT antigen function, specifically, viral replication [10,13,14][10][13][14]. MCV LT is a phosphoprotein and contains approximately 82 potential phosphorylation sites (p-sites), which are expected to be crucial for modulation of LT protein function [15]. Full-length LT initiates viral replication and induces Ataxia telangiectasia mutated (ATM) and Ataxia-telangiectasia- and Rad3-related (ATR)-mediated DNA damage response (DDR) and cell cycle arrest, leading to cell death [8,16,17][8][16][17]. It was previously observed that MCV LT serine (S) 816 phosphorylation by the ATM kinase stimulates DDR and cell growth inhibition [17]. Interestingly, an alanine (A) substitutional mutation of S816 only partly restores cell growth, suggesting the possibility of a secondary mechanism contributing to this underlying phenotype [17].
Growing evidence indicates that regulation of viral protein stability may be a key determinant for viral pathogenesis and oncogenesis in many human tumor viruses [18,19,20,21][18][19][20][21]. SV40 LT protein is relatively stable (t1/2 > 36 h), and regulated at lysine (K) 697 by a reversible acetylation reaction [22]. The effect of acetylation on SV40 LT stability is not changed by proteasomal inhibitor, MG132, indicating that lysine 697 acetylation has a predominant role in regulating SV40 LT stability. Moreover, Shimazu et al. described that a substitution mutation of K679 to glutamic acid (Q) reduced SV40 LT stability and cell growth, suggesting that LT stability is closely related to LT-mediated viral and host protein–protein interactions and cell proliferation.
MCV LT contains a unique domain that exhibits no homology to other polyomaviruses [23,24][23][24]. This region comprises two fragments: MCV unique region (MUR) 1 (106 aa) and MUR2 (39 aa), which flank the Rb binding motif [23,24][23][24]. This domain contains a unique vacuolar sorting protein (Vam6p) binding sequence (W209) [23], and phosphorylated serines (S147, S220, S239) that interact with Skp-Cullin-F-box (SCF) E3 ligases [15]. MCV LT interactions with SCF E3 ligase complexes induce self-destabilization [15], thus, we hypothesized that this domain functions as a negative regulatory element for LT-mediated cell growth. We sought to determine whether MCV LT MUR domain affects LT protein-mediated cell proliferation. Our results show that deletion of the MUR in MCV LT significantly increases MCV LT protein stability and cell proliferation due to loss of SCF E3 ligase interactions. In contrast, the insertion of MCV LT MUR sequences to SV40 LT destabilizes SV40 LT protein by enhancing SCF E3 ligase interactions, resulting in a moderate decrease in cell proliferation. Interestingly, upon deletion of the MUR domain, MCV LT retained its ability to replicate MCV viral origin and regulate LT-mediated MCV early gene transcription.
2. MCV LT Has a Unique and Disordered Domain Structure
The gene sequence of MCV LT is highly conserved among all MCV strains harboring an intact LT sequence [3]. We compared the aligned LT amino acid sequences of 14 human polyomaviruses using PLOTCON software (EMBOSS). As previously shown, the Rb-binding motif in MCV LT is located between the two unique regions (MUR1 and MUR2), unlike other polyomaviruses (Figure 1A) [23,24][23][24]. Further assessment of these sequences using IUPred2 and ANCHOR2 allowed us to investigate these MUR fragments, and we ascertained that these two regions are intrinsically disordered (IUPred2 analysis), which may contribute to the multi-functionality of LT including protein-protein interactions [29,30,31][25][26][27]. Because SV40 LT protein interactions and functions are well characterized, we compared sequence variations between SV40 LT and MCV LT (Figure 1B).
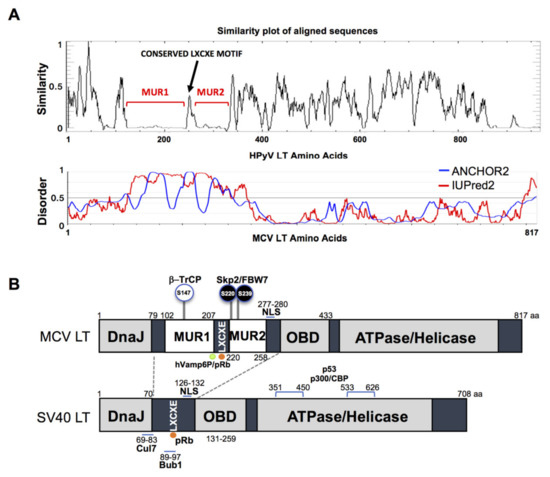
Figure 1. Merkel cell polyomavirus large tumor antigen (MCV LT) similarity and disorder plots. (A) LT amino acid similarity of 14 human polyomaviruses (HPyVs) and disordered region of MCV LT protein. Sliding window plot (6 aa window size) analysis. LT amino acid similarity was compared using PLOTCON (EMBOSS). For disordered region prediction, IUPred2 and ANCHOR2 were utilized to identify disordered protein regions and disordered binding regions in MCV LT, respectively. MCV LT contains a unique disordered region (MUR) divided in two fragments by the conserved LXCXE motif [23,24][23][24]. (B) Diagram of MCV LT and SV40 LT domain structures. Compared with SV40 LT, MCV LT has an extended structure, MUR, that serves as an interacting domain with multiple cellular factors. The MUR domain also consists of phosphorylated serines for SCF E3 ligase recognition (phospho-degron motifs) [15].
3. MCV LT Unique Region (MUR) Is Important for LT Stability
Previous studies determined that MCV LT MUR domains have multiple E3 ligase binding serines that are not present in SV40 LT [15,32][15][28]. To understand the effect of the MUR domains on MCV LT stability, we created a LT MUR deletion mutant (LTdMUR) (Figure 2A) and LT protein stability was analyzed in the presence and absence of the MUR domain using a CHX chase assay. As shown in Figure Figure 22B,C, deletion of the MUR domain significantly reduced LT protein turnover. To further verify if this stability phenotype was MUR-specific, we introduced the MUR sequences to SV40 LT (SV40 LT+MUR) (Figure 2D). Our analysis illustrated that introduction of the MUR mutation to SV40 LT significantly reduced protein stability as shown in Figure 2E. Immunoblot analysis demonstrated a significant reduction in SV40 LT half-life of 36 h [22] to approximately 6 h (Figure 2F).
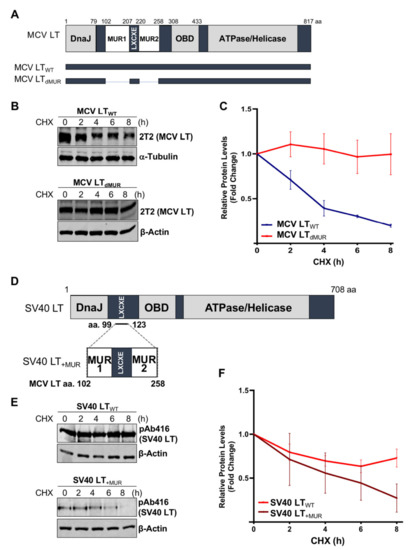
Figure 2. MCV LT unique region (MUR) regulates LT stability. (A) Diagram of MCV LT coding regions and sites of deletion mutations. MCV LT MUR domains: MUR1 (102–207 aa) and MUR2 (220–258 aa) were simultaneously deleted to evaluate LT stability. (B) LT MUR destabilizes LT. LT protein turnover was measured by a cycloheximide (CHX) chase assay using quantitative immunoblot analysis. Cells transfected with LTWT or LTdMUR constructs (0.3 µg and 0.9 µg respectively) were treated with CHX (0.1 mg/mL) 24 h after transfection and harvested at each time point indicated. (C) Protein expression was quantified using a LI-COR IR imaging system. Deletion of the MUR extended the half-life of LT from ~3–4 h up to >8 h. Error bars represent SEM and were calculated using GraphPad Prism software. Data were analyzed using three biological replicates per experiment, n = 3. (D) Diagram of SV40 LT coding regions and sites of insertion mutations. SV40 LT amino acids (99–123) were deleted, and the MCV LT complete MUR domain (102–258 aa) was inserted. (E) MCV LT MUR destabilizes SV40 LT. SV40 LT protein turnover was assessed by a CHX chase assay using quantitative immunoblot analysis. 293 cells transfected with either wild-type SV40 LT (SV40 LTWT) (0.3 µg) or SV40 LT+MUR (0.6 µg) were treated with CHX (0.1 mg/mL) 24 h after transfection and harvested at each time point indicated. (F) Protein expression was quantified using the laser-scanning Odyssey CLX (LI-COR) infrared (IR) imaging system. MCV MUR insertion into SV40 LT reduced SV40 LT turnover to ~6 h. Error bars represent standard errors of the mean (SEM) and were calculated using GraphPad Prism software. Data were analyzed using three biological replicates per experiment, n = 3.
References
- Becker, J.C.; Stang, A.; De Caprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Rev. Dis. Primers 2017, 3, 17077, doi:10.1038/nrdp.2017.77.
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100, doi:10.1126/science.1152586.
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Natl. Acad. Sci. USA 2008, 105, 16272–16277, doi:10.1073/pnas.0806526105.
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. Virol. 2009, 83, 12118–12128, doi:10.1128/JVI.01336-09.
- Harrison, C.J.; Meinke, G.; Kwun, H.J.; Rogalin, H.; Phelan, P.J.; Bullock, P.A.; Chang, Y.; Moore, P.S.; Bohm, A. Asymmetric assembly of Merkel cell polyomavirus large T-antigen origin binding domains at the viral origin. Mol. Biol. 2011, 409, 529–542, doi:10.1016/j.jmb.2011.03.051.
- Nakamura, T.; Sato, Y.; Watanabe, D.; Ito, H.; Shimonohara, N.; Tsuji, T.; Nakajima, N.; Suzuki, Y.; Matsuo, K.; Nakagawa, H.; et al. Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology 2010, 398, 273–279, doi:10.1016/j.virol.2009.12.024.
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; De Caprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. Virol. 2013, 87, 6118–6126, doi:10.1128/JVI.00385-13.
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. Virol. 2013, 87, 9173–9188, doi:10.1128/JVI.01216-13.
- Sacco, F.; Perfetto, L.; Castagnoli, L.; Cesareni, G. The human phosphatase interactome: An intricate family portrait. FEBS Lett. 2012, 586, 2732–2739, doi:10.1016/j.febslet.2012.05.008.
- Chatterjee, A.; Bockus, B.J.; Gjorup, O.V.; Schaffhausen, B.S. Phosphorylation sites in polyomavirus large T antigen that regulate its function in viral, but not cellular, DNA synthesis. Virol. 1997, 71, 6472–6478, doi:10.1128/jvi.71.9.6472-6478.1997.
- Hassauer, M.; Scheidtmann, K.H.; Walter, G. Mapping of phosphorylation sites in polyomavirus large T-antigen. Virol. 1986, 58, 805–816, doi:10.1128/jvi.58.3.805-816.1986.
- Fluck, M.M.; Schaffhausen, B.S. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Mol. Biol. Rev. 2009, 73, 542–563, doi:10.1128/mmbr.00009-09.
- Prives, C. The replication functions of SV40 T-antigen are regulated by phosphorylation. Cell 1990, 61, 735–738, doi:10.1016/0092-8674(90)90179-i.
- McVey, D.; Brizuela, L.; Mohr, I.; Marshak, D.R.; Gluzman, Y.; Beach, D. Phosphorylation of large tumor-antigen by cdc2 stimulates SV40 DNA-replication. Nature 1989, 341, 503–507, doi:10.1038/341503a0.
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-Mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Natl. Acad. Sci. USA 2017, 114, E4040–E4047, doi:10.1073/pnas.1703879114.
- Tsang, S.H.; Wang, X.; Li, J.; Buck, C.B.; You, J. Host DNA damage response factors localize to merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. Virol. 2014, 88, 3285–3297, doi:10.1128/JVI.03656-13.
- Li, J.; Diaz, J.; Wang, X.; Tsang, S.H.; You, J. Phosphorylation of Merkel cell polyomavirus large tumor antigen at serine 816 by ATM kinase induces apoptosis in host cells. Biol. Chem. 2015, 290, 1874–1884, doi:10.1074/jbc.M114.594895.
- Chen, J.; Wu, X.; Chen, S.; Xiang, N.; Chen, Y.; Guo, D. Ubiquitin ligase Fbw7 restricts the replication of hepatitis C virus by targeting NS5B for ubiquitination and degradation. Biophys. Res. Commun. 2016, 470, 697–703, doi:10.1016/j.bbrc.2016.01.076.
- Alvarez Orellana, J.; Kwun, H.J.; Artusi, S.; Chang, Y.; Moore, P.S. Sirolimus and other mTOR inhibitors directly activate latent pathogenic human polyomavirus replication. Infect. Dis. Jiaa071, 2020, doi:10.1093/infdis/jiaa071.
- Bellanger, S.; Tan, C.L.; Nei, W.; He, P.P.; Thierry, F. The human papillomavirus type 18 E2 protein is a cell cycle-dependent target of the SCFSkp2 ubiquitin ligase. Virol. 2010, 84, 437–444, doi:10.1128/JVI.01162-09.
- Hou, L.; Zhao, J.; Gao, S.; Ji, T.; Song, T.; Li, Y.; Wang, J.; Geng, C.; Long, M.; Chen, J.; et al. Restriction of hepatitis B virus replication by c-Abl-induced proteasomal degradation of the viral polymerase. Adv. 2019, 5, eaau7130, doi:10.1126/sciadv.aau7130.
- Shimazu, T.; Komatsu, Y.; Nakayama, K.I.; Fukazawa, H.; Horinouchi, S.; Yoshida, M. Regulation of SV40 large T-antigen stability by reversible acetylation. Oncogene 2006, 25, 7391–7400, doi:10.1038/sj.onc.1209731.
- Liu, X.; Hein, J.; Richardson, S.C.; Basse, P.H.; Toptan, T.; Moore, P.S.; Gjoerup, O.V.; Chang, Y. Merkel cell polyomavirus large T antigen disrupts lysosome clustering by translocating human Vam6p from the cytoplasm to the nucleus. Biol. Chem. 2011, 286, 17079–17090, doi:10.1074/jbc.M110.192856.
- Houben, R.; Angermeyer, S.; Haferkamp, S.; Aue, A.; Goebeler, M.; Schrama, D.; Hesbacher, S. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int. J. Cancer 2015, 136, E290–E300, doi:10.1002/ijc.29200.
References
- Boutayeb, A. The impact of infectious diseases on the development of Africa. In The Handbook of Disease Burdens and Quality of Life Measures; Preedy, V.R., Watson, R.R., Eds.; Springer: New York, NY, USA, 2010; pp. 1171–1188, ISBN 978-0-387-78664-3.
- Kuupiel, D.; Bawontuo, V.; Drain, P.K.; Gwala, N.; Mashamba-Thompson, T.P. Supply chain management and accessibility to point-of-care testing in resource-limited settings: A systematic scoping review. BMC Heal. Serv. Res. 2019, 19, 519, doi:10.1186/s12913-019-4351-3.
- Starr, A.; Graef, K.M.; Dent, J. Fostering innovative product development for neglected tropical diseases through partnerships. Pharm. Pat. Anal. 2016, 5, 391–400, doi:10.4155/ppa-2016-0038.
- Migliozzi, D.; Guibentif, T. Assessing the potential deployment of biosensors for point-of-care diagnostics in developing countries: Technological, economic and regulatory aspects. Biosensors 2018, 8, 119, doi:10.3390/bios8040119.
- Pai, N.P.; Vadnais, C.; Denkinger, C.; Engel, N.; Pai, M. Point-of-care testing for infectious diseases: Diversity, complexity, and barriers in low-and middle-income countries. PLoS Med. 2012, 9, e1001306, doi:10.1371/journal.pmed.1001306.
- Sia, S.K.; Kricka, L.J. Microfluidics and point-of-care testing. Lab Chip 2008, 8, 1982–1983, doi:10.1039/B817915H.
- Kozel, T.R.; Burnham-Marusich, A.R. Point-of-care testing for infectious diseases: Past, present, and future. J. Clin. Microbiol. 2017, 55, 2313–2320, doi:10.1128/jcm.00476-17.
- Pashchenko, O.; Shelby, T.; Banerjee, T.; Santra, S.A. Comparison of optical, electrochemical, magnetic, and colorimetric point-of-care biosensors for infectious disease diagnosis. ACS Infect. Dis. 2018, 4, 1162–1178, doi:10.1021/acsinfecdis.8b00023.
- Lowdon, J.W.; Eersels, K.; Rogosic, R.; Boonen, T.; Heidt, B.; Diliën, H.; Van Grinsven, B.; Cleij, T.J. Surface grafted molecularly imprinted polymeric receptor layers for thermal detection of the new psychoactive substance 2-methoxphenidine. Sens. Actuators. 2019, 295, 586–595, doi:10.1016/j.sna.2019.06.029.
- Vandenryt, T.; Van Grinsven, B.; Eersels, K.; Cornelis, P.; Kholwadia, S.; Cleij, T.J.; Thoelen, R.; De Ceuninck, W.; Peeters, M.; Wagner, P. Single-shot detection of neurotransmitters in whole-blood samples by means of the heat-transfer method in combination with synthetic receptors. Sensors 2017, 17, 2701, doi:10.3390/s17122701.
- Takemura, K.; Adegoke, O.; Takahashi, N.; Kato, T.; Li, T.-C.; Kitamoto, N.; Tanaka, T.; Suzuki, T.; Park, E.Y. Versatility of a localized surface plasmon resonance-based gold nanoparticle-alloyed quantum dot nanobiosensor for immunofluorescence detection of viruses. Biosens. Bioelectron. 2017, 89, 998–1005, doi:10.1016/j.bios.2016.10.045.
- Lowdon, J.W.; Eersels, K.; Rogosic, R.; Heidt, B.; Diliën, H.; Redeker, E.S.; Peeters, M.; Van Grinsven, B.; Cleij, T.J. Substrate displacement colorimetry for the detection of diarylethylamines. Sens. Actuators. 2019, 282, 137–144, doi:10.1016/j.snb.2018.11.053.
- Xiong, L.-H.; Cui, R.; Zhang, Z.-L.; Yu, X.; Xie, Z.; Shi, Y.-B.; Pang, D.-W. Uniform fluorescent nanobioprobes for pathogen detection. ACS Nano 2014, 8, 5116–5124, doi:10.1021/nn501174g.
- Myers, F.B.; Lee, L.P. Innovations in optical microfluidic technologies for point-of-care diagnostics. Lab Chip 2008, 8, 2015–2031, doi:10.1039/b812343h.
- Afsahi, S.; Lerner, M.B.; Goldstein, J.M.; Lee, J.; Tang, X.; Bagarozzi, D.A.; Pan, D.; Locascio, L.; Walker, A.; Barron, F.E.; et al. Novel graphene-based biosensor for early detection of Zika virus infection. Biosens. Bioelectron. 2018, 100, 85–88, doi:10.1016/j.bios.2017.08.051.
- Campuzano, S.; Yáñez-Sedeño, P.; Pingarrón, J. M. Molecular biosensors for electrochemical detection of infectious pathogens in liquid biopsies: Current trends and challenges. Sensors 2017, 17, 2533, doi:10.3390/s17112533.
- Cecchetto, J.; Fernandes, F.C.B.; Lopes, R.; Bueno, P.R. The capacitive sensing of NS1 Flavivirus biomarker. Biosens. Bioelectron. 2017, 87, 949–956, doi:10.1016/j.bios.2016.08.097.
- Hsieh, K.; Ferguson, B.S.; Eisenstein, M.; Plaxco, K.W.; Soh, H.T. Integrated electrochemical microsystems for genetic detection of pathogens at the point of care. Acc. Chem. Res. 2015, 48, 911–920, doi:10.1021/ar500456w.
- Park, J.Y.; Kricka, L.J. Prospects for nano-and microtechnologies in clinical point-of-care testing. Lab Chip 2007, 7, 547–549, doi:10.1039/b702667f.
- Kuupiel, D.; Bawontuo, V.; Mashamba-Thompson, T.P. Improving the accessibility and efficiency of point-of-care diagnostics services in low-and middle-income countries: Lean and agile supply chain management. Diagnostics 2017, 7, 58, doi:10.3390/diagnostics7040058.
- Wang, S.; Lifson, M.A.; Inci, F.; Liang, L.-G.; Sheng, Y.-F.; Demirci, U. Advances in addressing technical challenges of point-of-care diagnostics in resource-limited settings. Expert Rev. Mol. Diagn. 2016, 16, 449–459, doi:10.1586/14737159.2016.1142877.
- Urdea, M.; Penny, L.A.; Olmsted, S.S.; Giovanni, M.Y.; Kaspar, P.; Shepherd, A.; Wilson, P.; Dahl, C.A.; Buchsbaum, S.; Moeller, G.; et al. Requirements for high impact diagnostics in the developing world. Nature 2006, 444, (Suppl 1), 73–79, doi:10.1038/nature05448.
- Tayoun, A.N.A.; Ward, B.P.; Maltezos, G.; Scherer, A.; Tsongalis, G.J. Evaluating the thermostability of commercial fast real-time PCR master mixes. Exp. Mol. Pathol. 2012, 93, 261–263, doi:10.1016/j.yexmp.2012.05.002.
- Furuse, Y. Analysis of research intensity on infectious disease by disease burden reveals which infectious diseases are neglected by researchers. Proc. Natl. Acad. Sci. USA 2019, 116, 478–483, doi:10.1073/pnas.1814484116.
- Expedited Programs for Serious Conditions––Drugs and Biologics. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expedited-programs-serious-conditions-drugs-and-biologics (accessed on 9 July 2020)
- Ridley, D.B.; Régnier, S.A. The commercial market for priority review vouchers. Health Aff. 2016, 35, 776–783, doi:10.1377/hlthaff.2015.1314.
- Slingsby, B.; Kurokawa, K. The Global Health Innovative Technology (GHIT) Fund: Financing medical innovations for neglected populations. Lancet Glob. Health 2013, 1, e184–e185, doi:10.1016/s2214-109x(13)70055-x.
- Bessa, T.C.B.; De Aragão, E.S.; Guimarães, J.M.M.; Almeida, B.D.A. R&D in vaccines targeting neglected diseases: An exploratory case study considering funding for preventive tuberculosis vaccine development from 2007 to 2014. Biomed Res. Int. 2017, 2017, 4765719, doi:10.1155/2017/4765719.