Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Eulàlia Genescà.
Thanks to the use of high-resolution genetic techniques to detect cryptic aberrations present in T-ALL, wresearchers now have a clearer view of the genetic landscape that explains the particular oncogenetic processes taking place in each T-ALL. WeThey also have begun to understand relapse-specific mechanisms. This review aims to summarize the latest advances in ourthe knowledge of the genome in T-ALL and highlight the areas where the research on this ALL subtype is progressing, thereby identifying the key issues that need to be addressed in the medium-to-long term to move forward in the applicability of this knowledge into clinics.
- T-cell acute lymphoblastic leukemia
- genomics
1. Introduction
ALL includes B- and T-cell lineage subtypes. While these diseases share many similarities, such as some general cancer genetic lesions in cell cycle regulators such as CDKN2A/B and MYC [2][1], their genetic background, origin, and outcome are distinct. The T-ALL subtype accounts for 10–15% of pediatric and 25% of adult ALL cases. It is more frequent in males than in females, and more often presents at younger ages, being considered an AYA (adolescent and young adult)-affecting disease [3][2]. Although intensive chemotherapy treatments have been crucial for increasing the survival rate of affected children up to 93% [4][3], globally, adult survival rates are still very poor, remaining below 50% [5][4]. The main cause of treatment failure and worse outcome is relapse [6,7][5][6]. In this scenario, T-ALL patients with primary-resistant or relapsed leukemia present a dismal prognosis, mainly due to the rapid progression of the recurrent course of the disease and the lack of effective therapeutic options [7,8][6][7].
Initial genetic studies based on the use of cytogenetics and FISH helped identify some recurrent chromosomal alterations in T-ALL at the time of diagnosis, but it proved difficult to determine their prognostic impact because of their low incidence in the specific T-ALL cohort analyzed. Genetic knowledge flourished with the application of genomic techniques, first with the analysis of gene expression profiles (GEPs), followed by the application of comparative genomic hybridization arrays (CGHas), and later, the next generation sequencing (NGS) technique. Consequently, wresearchers now have a clearer appreciation of the different T-ALL genetic subtypes at the time of diagnosis and are beginning to understand relapse-specific mechanisms.
2. T-ALL Classification by Differentiation Stage
The main contribution of genomic techniques in T-ALL has been to show that the blockade of the differentiation process that occurs in the lymphocyte is the consequence of specific genetic alterations occurring in pre-leukemic stages. From a historical perspective, the first studies to classify T-ALL aroused with the use of the gene expression array (GEa). This revealed that structural abnormalities, mainly rearrangements identified in T-ALL by karyotyping as a rare event [9,10,11,12[8][9][10][11][12][13][14][15][16][17][18],13,14,15,16,17,18,19], led to much overexpression of (1) basic helix-loop-helix (bHLH) transcription factor genes such as TAL1, TAL2, and LYL1; (2) LIM-only (LMO) domain genes such as LMO1 and LMO2; and (3) homeobox genes such as TLX1 (HOX11) and TLX3 (HOX11L2). Supervised analysis of the expression data revealed a correlation between the expression of these transcription factors (TFs) and a lymphocyte-specific differentiation arrest time point. Three main groups (clusters) were obtained: (1) immature, (2) HOXA, and (3) TAL [20,21][19][20]. To describe this in more detail, HOXA samples cluster with the Pre-T1 (CD34 + CD1a+) and Pre-T2/Pre-T3 (CD4+, CD8αβ-, CD3- and TCR αβ-) sub-populations, together with the TLX1-expressing cases and some cases expressing TLX3. TAL samples cluster with subpopulations corresponding to thymocytes with a pre-TCR (Beta selection). Of note, samples from the immature group included some TLX3-expressing leukemic samples and clustered with the most immature early T-cell precursor and pro-T subpopulations [21][20]. Later on, the addition of copy number data, generated by CGHa, together with the use of NGS, yielded a clearer view of the genetic determinants that define these groups. Thus, according to the differentiation arrest time point of the blast cell wresearchers can differentiate the immature subtype, characterized by the absence of CD8 and CD1a immunomarkers, high levels of expression of LYL1 [20][19] and MEF2C [22][21] TFs, and the absence of bi-allelic deletions in TCRγ [23][22]. Within the immature T-ALL leukemias, the early T-cell precursor ALL (ETP-ALL)—defined by the absence of CD1a and CD8 immunomarkers and the presence of stem cells or myeloid markers such as CD117, CD34, HLA-DR, CD13, CD33, CD11b, and CD65 [24][23], together with negative or dim CD5 expression, defined as expression in <75% of the blasts; rarely presents CDKN2A/B deletions and NOTCH1/FBXW7 mutations [25,26,27][24][25][26]. In addition, mutations affecting epigenetic regulators and transcription factors governing hematopoietic and T-cell development are also frequently observed in this immature subtype [25,28,29,30,31][24][27][28][29][30]. WResearchers will discuss this subgroup in detail later. The cortical subtype is characterized by the expression of CD1a, and often both CD4 and CD8 immunomarkers are also found. At the genetic level, the group is characterized by aberrant expression of TLX1, TLX3, and HOXA genes (HOXA5, HOXA9, HOXA10) [20,21][19][20] and the overexpression of the NKX2-1 rearranged gene [22][21]. Other specific alterations in this subset of T-ALL have been found in PHF6, DNM2, BCL11B, CDKN1B, and RB1 [32][31]. The mature subtype is characterized by blasts expressing CD4 or CD8 and surface CD3 immunomarkers [20][19] and the presence of Tαβ cell-receptor rearrangements [21][20]. The genetic hallmark of the group is the activation of the TAL oncogene [20][19], together with the presence of del(6)(q) [33][32] and mutations in PIK3R and PTEN [32][31]. Figure 1 summarizes these findings.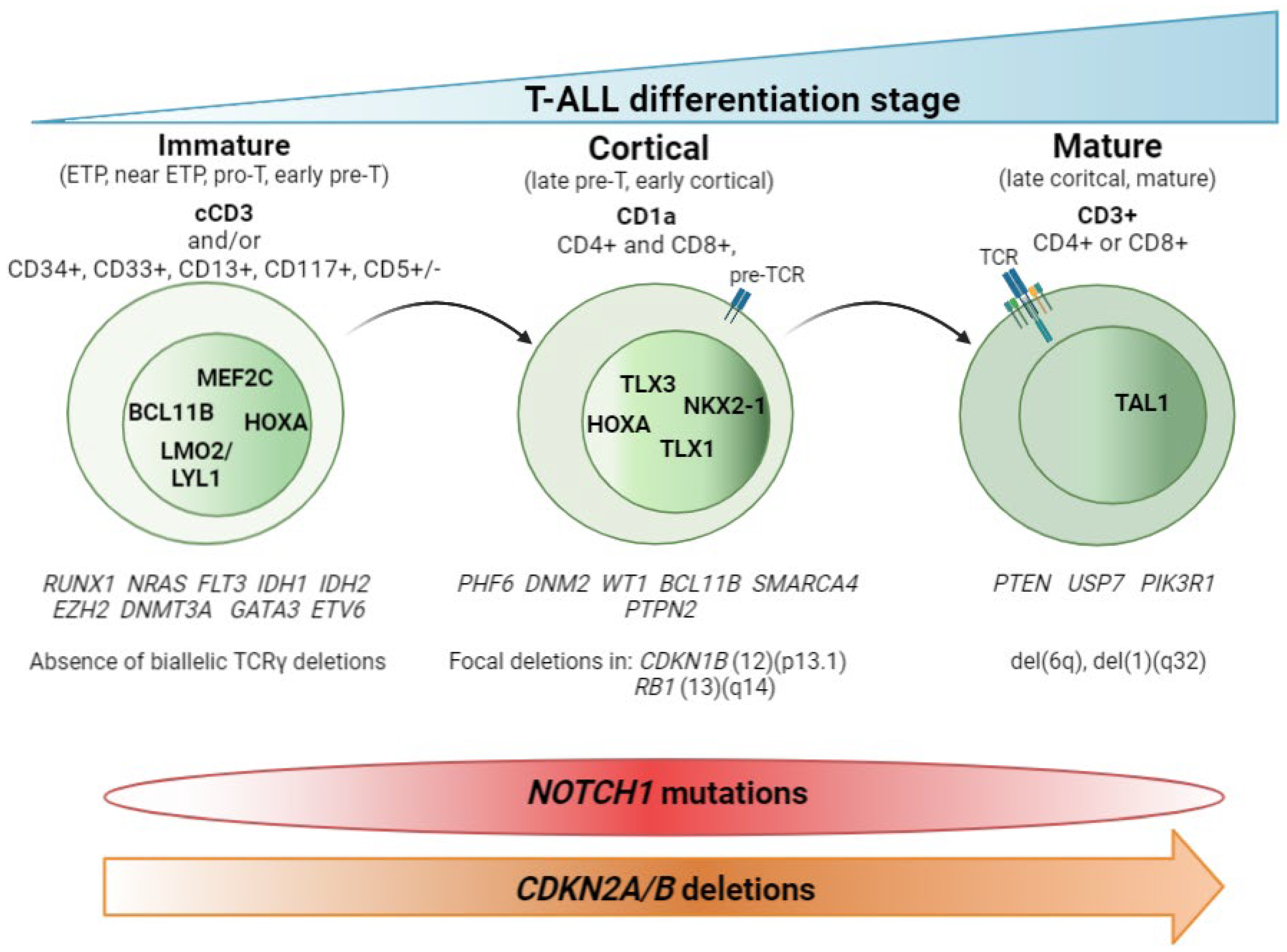
Figure 1. T-ALL classification by stage of differentiation arrest. Schematic representation of the three main T-ALL subtypes according to the blockade of the differentiation process. Hallmark immunomarkers of each subtype are highlighted in bold at the top. Other accompanying immunomarkers, for precise definition of the subtypes, are showed below. TCR maturation is represented in the blasts. Presence of TCR on the blast surface is also a hallmark in the mature subtype. Active transcription factors in each subtype of T-ALL are represented in the nucleus according to the maturation transition. The genes most frequently mutated in each subtype, followed by copy number alterations, are shown beneath. The distributions of NOTCH1 mutations and CDKN2A/B deletions are indicated at the bottom.
2.1. Non-Coding Mutations
Analysis of non-coding data generated by whole genome sequencing (WGS) or direct target sequencing is an emerging area of investigation, but recently published data have shown that small (or not) insertions and deletions generating novel regulatory sequences can explain the overexpression seen in TFs, such as TAL1 and LMO1, that have no detectable primary rearrangements. Here wresearchers provide a summary of the non-coding alterations identified in TAL1, LMO1/2, and other important T-ALL oncogenes such as MYC and PTEN. Non-coding variants discovered in T-ALL are also summarized in Table 1.Table 1. Non-coding mutations identified so far in T-ALL.
| Gene | Affected Region |
Variant | Alteration | Functional Impact |
Frequency | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MYC | 1427 kb downstream of | MYC | Focal duplications |
Creation of binding site for | NOTCH1 | MYC | expression |
8/160 (5%) Adult and pediatric |
[34] | [33] | ||
| TAL1 | 8 kb upstream of the transcription start site of | TAL1 | Heterozygous indel (2–18 bp) | Creation of binding motifs for the | MYB | TF | TAL1 | overexpression |
8/146 (5.5%) pediatric |
[35,36] | [34][35] | |
| LMO1 | 4 kb upstream of the transcriptional start site of | LMO1 | SNV: C → T | Creation of binding motifs for the | MYB | TF | LMO1 | overexpression |
4/187 (2.14%) pediatric | [36,37] | [35][36] | |
| LMO2 | Non-coding region of the exon 2 of | LMO2 | Heterozygous indel |
Creation of binding motifs for the | MYB | TF | Activating | LMO2 | function | 6/160 (3.75%) pediatric 9/163 (5.52%) adult |
[38] | [37] |
| PTEN | 550 kb downstream of transcription start site of | PTEN | Focal deletions | Deletion of | PTEN | enhancer region | Reduced levels of | PTEN | 5/398 (1.25%) | [39] | [38] |
2.2. T-ALL Related Immature Subtypes
Application of WGS and whole transcriptome sequencing has also served to better characterize rare subtypes such as the immature T-ALL leukemias and to provide insight into the cell of origin of these subtypes. This group includes T/Myeloid mixed phenotype acute leukemias (T/M MPALs) and ETP-ALL, which are characterized by different combinations of myeloid and T-lymphoid antigen expression [40][39]. Other immunophenotypically identified immature T-ALL subtypes include the pro-T [40][39] and the near-ETP [32,41][31][40] forms, but wresearchers do not know their genetic basis or clinical implications. Thus, childhood ETP-ALL presents cytokine-activating somatic mutations and mutations in genes involved in the RAS signaling pathway (e.g., NRAS, KRAS, FLT3, IL7R, JAK3, JAK1, SH2B3, and BRAF), genetic alterations that inactivate genes involved in hematopoietic development (e.g., GATA3, ETV6, RUNX1, IKZF1, and EP300), and mutations in histone modifier genes (e.g., EZH2, EED, SUZ12, SETD2, and EP300). It is of note that the mutational spectrum identified in ETP leukemia was similar to that of acute myeloid leukemia (AML) with poor prognosis, in which affected pluripotent genes lend this subtype a myeloid-like profile [29][28]. In the case of adult ETP-ALL, exclusively genetic alterations have been detected in the DNMT3A gene (frequency range from 12 to 16%) [30[29][30][41][42],31,42,43], in addition to the aforementioned mutations. Specifically, DNMT3A mutations are associated with patients aged >60 years with ETP-ALL features [43][42]. FAT1 (25%, 17/68) and FAT3 (20%, 14/68) cadherins are other mutations exclusively found in adult ETP-ALL [30][29] (Figure 2). In addition to point mutations, structural abnormalities such as rearrangements affecting KMT2A, MLLT10, NUP214, or NUP98, which trans activate HOXA genes, are often detected in ETP-ALL cases [44,45][43][44]. Overexpression of the BCL11B gene due to different structural abnormalities including translocations (i.e., t(2;14)(q22.3;q32), t(6;14)(q25.3;q32), hijacking super-enhancers, and other fusion genes has been very recently described as present in one third of ETP-ALL and T/myeloid mixed phenotype acute leukemia (T/M MPAL) cases with a very distinct expression profile [46,47][45][46].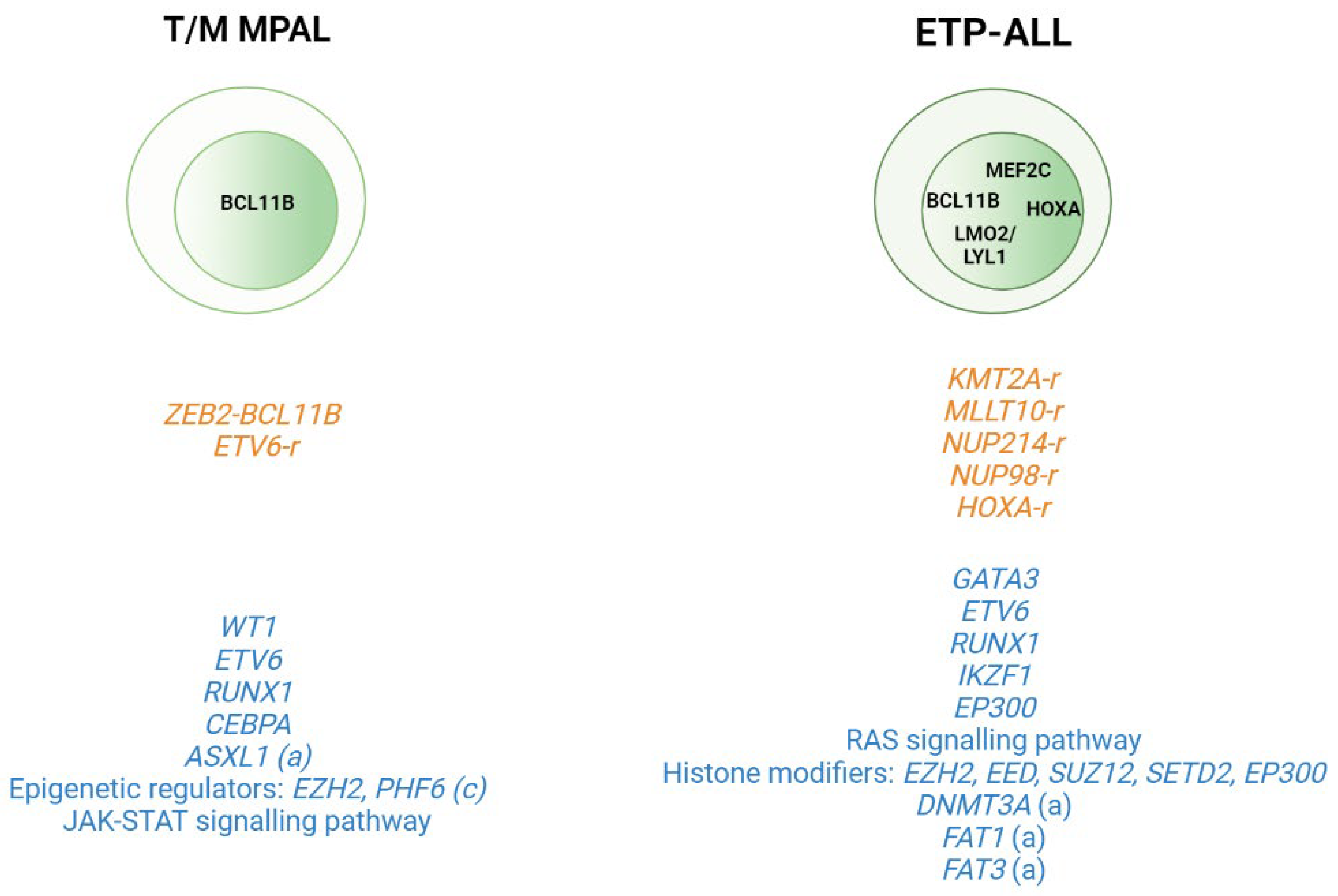
Figure 2. Genomic alterations in T/Myeloid mixed phenotype acute leukemias (T/M MPALs) and ETP-ALL. Active transcription factors in each subtype are represented in the nucleus according to the maturation transition. The rearrangements and fusion genes are written in yellow and most frequently mutations in blue color. (a) adult; (c) children.
3. (Epi)genetic Modification
The systematic screening of T-ALL genomes has revealed T-ALL as one of the tumors with the highest frequency of mutations in genes that encode proteins involved in epigenetic regulation [50][49]. Hence, the field of epigenetics, particularly DNA methylation, is currently being extensively explored in the search for specific methylation patterns that help to explain the oncogenic evolution of pre-leukemic T-cells; to identify specific de-regulated genes to use as a prognosis marker; and to delineate new therapy strategies using DNA methylation inhibitors (iDNMTs) such as 5-azacitidine (vidaza, AZA) and 5-aza-20 -deoxycytidine (decitabine, DAC) [51][50]. Initial epigenetic studies were focused on determining the methylation status of the promoter of specific genes playing a role in the T-ALL oncogenic process such as CDKN2A/B. It was observed in T-ALL patients that the percentage of promoter methylation in the CDKN2B and CDKN2A genes ranged between 46% and 68% and between 0% and 12%, respectively, in pediatric cohorts ([52,53,54][51][52][53]. In the case of adult T-ALL cohorts, the percentage of CDKN2B gene promoter methylation varied from 16% to 49% and was 1% for the CDKN2A promoter [55,56,57,58,59][54][55][56][57][58]. In T-ALL, the CDKN2B methylation status was associated with an immature immunophenotype [58][57] and with ETP-ALL features [59][58]. Further investigation of the methylation status in cancer cells using wide genomic approaches (i.e., methylation arrays) have observed that generally malignant cells display a DNA hypermethylation pattern at specific CpG islands; globally, this observation is called a CpG island methylator phenotype (CIMP). CIMP+ in T-ALLs has been associated with a better EF and OS as compared to CIMP− leukemias [60][59]. Notably, these findings have been confirmed in both pediatric [61][60] and adult [62][61] T-ALL cohorts, reinforcing the idea that aberrant DNA methylation might act as a clinically relevant biomarker in human T-ALL. Comparison of the global methylation profile of T-ALL samples with that of normal thymocytes observed that the methylation profile of CIMP− cases was close to normal CD3+ and CD34+ thymocytes [60,61][59][60]. That was interpreted as an indication of a shorter proliferation history of the CIMP− blasts as compared to CIMP+ cases [63][62]. Together, these findings indicate that CIMP− cases are characterized by a hypomethylation pattern that results in a young mitotic age and shorter proliferative history of leukemic cells; at the same time, however, it might be considered as a marker of higher aggressiveness of leukemic cells. In CIMP+ cases, the disease latency is longer, which is reflected by higher methylation acquired during the aging of pre-leukemic cells, [61,63,64][60][62][63]. Altogether, these results indicated that aberrant methylation is likely not a driving force of T-ALL onset and progression but is rather related to the proliferative history of the cells. These concepts have been nicely reviewed by Natalia Mackowska et al. [65][64] in a recent publication.References
- Belver, L.; Ferrando, A. The Genetics and Mechanisms of T Cell Acute Lymphoblastic Leukaemia. Nat. Rev. Cancer 2016, 16, 494–507.
- Guru Murthy, G.S.; Pondaiah, S.K.; Abedin, S.; Atallah, E. Incidence and Survival of T-Cell Acute Lymphoblastic Leukemia in the United States. Leuk. Lymphoma 2019, 60, 1171–1178.
- Bartram, J.; Veys, P.; Vora, A. Improvements in Outcome of Childhood Acute Lymphoblastic Leukaemia (ALL) in the UK—A Success Story of Modern Medicine through Successive UKALL Trials and International Collaboration. Br. J. Haematol. 2020, 191, 562–567.
- Ribera, J.-M.; Morgades, M.; Ciudad, J.; Montesinos, P.; Esteve, J.; Genescà, E.; Barba, P.; Ribera, J.; García-Cadenas, I.; Moreno, M.J.; et al. Chemotherapy or Allogeneic Transplantation in High-Risk Philadelphia Chromosome-Negative Adult Lymphoblastic Leukemia. Blood 2021, 137, 1879–1894.
- Beldjord, K.; Chevret, S.; Asnafi, V.; Huguet, F.; Boulland, M.-L.; Leguay, T.; Thomas, X.; Cayuela, J.-M.; Grardel, N.; Chalandon, Y.; et al. Oncogenetics and Minimal Residual Disease Are Independent Outcome Predictors in Adult Patients with Acute Lymphoblastic Leukemia. Blood 2014, 123, 3739–3749.
- Nguyen, K.; Devidas, M.; Cheng, S.-C.; La, M.; Raetz, E.A.; Carroll, W.L.; Winick, N.J.; Hunger, S.P.; Gaynon, P.S.; Loh, M.L. Factors Influencing Survival After Relapse From Acute Lymphoblastic Leukemia: A Children’s Oncology Group Study. Leukemia 2008, 22, 2142–2150.
- Barba, P.; Morgades, M.; Montesinos, P.; Gil, C.; Fox, M.-L.; Ciudad, J.; Moreno, M.-J.; González-Campos, J.; Genescà, E.; Martínez-Carballeira, D.; et al. Increased Survival Due to Lower Toxicity for High-Risk T-Cell Acute Lymphoblastic Leukemia Patients in Two Consecutive Pediatric-Inspired PETHEMA Trials. Eur. J. Haematol. 2019, 102, 79–86.
- Begley, C.G.; Aplan, P.D.; Davey, M.P.; Nakahara, K.; Tchorz, K.; Kurtzberg, J.; Hershfield, M.S.; Haynes, B.F.; Cohen, D.I.; Waldmann, T.A. Chromosomal Translocation in a Human Leukemic Stem-Cell Line Disrupts the T-Cell Antigen Receptor Delta-Chain Diversity Region and Results in a Previously Unreported Fusion Transcript. Proc. Natl. Acad. Sci. USA 1989, 86, 2031–2035.
- Bernard, O.; Guglielmi, P.; Jonveaux, P.; Cherif, D.; Gisselbrecht, S.; Mauchauffe, M.; Berger, R.; Larsen, C.J.; Mathieu-Mahul, D. Two Distinct Mechanisms for the SCL Gene Activation in the t(1;14) Translocation of T-Cell Leukemias. Genes Chromosom. Cancer 1990, 1, 194–208.
- Chen, Q.; Cheng, J.T.; Tasi, L.H.; Schneider, N.; Buchanan, G.; Carroll, A.; Crist, W.; Ozanne, B.; Siciliano, M.J.; Baer, R. The Tal Gene Undergoes Chromosome Translocation in T Cell Leukemia and Potentially Encodes a Helix-Loop-Helix Protein. EMBO J. 1990, 9, 415–424.
- Xia, Y.; Brown, L.; Yang, C.Y.; Tsan, J.T.; Siciliano, M.J.; Espinosa, R.; Le Beau, M.M.; Baer, R.J. TAL2, a Helix-Loop-Helix Gene Activated by the (7;9)(Q34;Q32) Translocation in Human T-Cell Leukemia. Proc. Natl. Acad. Sci. USA 1991, 88, 11416–11420.
- Mellentin, J.D.; Smith, S.D.; Cleary, M.L. Lyl-1, a Novel Gene Altered by Chromosomal Translocation in T Cell Leukemia, Codes for a Protein with a Helix-Loop-Helix DNA Binding Motif. Cell 1989, 58, 77–83.
- McGuire, E.A.; Hockett, R.D.; Pollock, K.M.; Bartholdi, M.F.; O’Brien, S.J.; Korsmeyer, S.J. The t(11;14)(P15;Q11) in a T-Cell Acute Lymphoblastic Leukemia Cell Line Activates Multiple Transcripts, Including Ttg-1, a Gene Encoding a Potential Zinc Finger Protein. Mol. Cell Biol. 1989, 9, 2124–2132.
- Boehm, T.; Foroni, L.; Kaneko, Y.; Perutz, M.F.; Rabbitts, T.H. The Rhombotin Family of Cysteine-Rich LIM-Domain Oncogenes: Distinct Members Are Involved in T-Cell Translocations to Human Chromosomes 11p15 and 11p13. Proc. Natl. Acad. Sci. USA 1991, 88, 4367–4371.
- Royer-Pokora, B.; Loos, U.; Ludwig, W.D. TTG-2, a New Gene Encoding a Cysteine-Rich Protein with the LIM Motif, Is Overexpressed in Acute T-Cell Leukaemia with the t(11;14)(P13;Q11). Oncogene 1991, 6, 1887–1893.
- Dube, I.; Kamel-Reid, S.; Yuan, C.; Lu, M.; Wu, X.; Corpus, G.; Raimondi, S.; Crist, W.; Carroll, A.; Minowada, J. A Novel Human Homeobox Gene Lies at the Chromosome 10 Breakpoint in Lymphoid Neoplasias with Chromosomal Translocation t(10;14). Blood 1991, 78, 2996–3003.
- Hatano, M.; Roberts, C.W.; Minden, M.; Crist, W.M.; Korsmeyer, S.J. Deregulation of a Homeobox Gene, HOX11, by the t(10;14) in T Cell Leukemia. Science 1991, 253, 79–82.
- Bernard, O.A.; Busson-LeConiat, M.; Ballerini, P.; Mauchauffé, M.; Della Valle, V.; Monni, R.; Nguyen Khac, F.; Mercher, T.; Penard-Lacronique, V.; Pasturaud, P.; et al. A New Recurrent and Specific Cryptic Translocation, t(5;14)(Q35;Q32), Is Associated with Expression of the Hox11L2 Gene in T Acute Lymphoblastic Leukemia. Leukemia 2001, 15, 1495–1504.
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene Expression Signatures Define Novel Oncogenic Pathways in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2002, 1, 75–87.
- Soulier, J.; Clappier, E.; Cayuela, J.-M.; Regnault, A.; García-Peydró, M.; Dombret, H.; Baruchel, A.; Toribio, M.-L.; Sigaux, F. HOXA Genes Are Included in Genetic and Biologic Networks Defining Human Acute T-Cell Leukemia (T-ALL). Blood 2005, 106, 274–286.
- Homminga, I.; Pieters, R.; Langerak, A.W.; de Rooi, J.J.; Stubbs, A.; Verstegen, M.; Vuerhard, M.; Buijs-Gladdines, J.; Kooi, C.; Klous, P.; et al. Integrated Transcript and Genome Analyses Reveal NKX2-1 and MEF2C as Potential Oncogenes in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2011, 19, 484–497.
- Gutierrez, A.; Dahlberg, S.E.; Neuberg, D.S.; Zhang, J.; Grebliunaite, R.; Sanda, T.; Protopopov, A.; Tosello, V.; Kutok, J.; Larson, R.S.; et al. Absence of Biallelic TCRγ Deletion Predicts Early Treatment Failure in Pediatric T-Cell Acute Lymphoblastic Leukemia. JCO 2010, 28, 3816–3823.
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-Cell Precursor Leukaemia: A Subtype of Very High-Risk Acute Lymphoblastic Leukaemia. Lancet Oncol. 2009, 10, 147–156.
- Van Vlierberghe, P.; Ambesi-Impiombato, A.; De Keersmaecker, K.; Hadler, M.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; Forne, C.; Rue, M.; Ferrando, A.A. Prognostic Relevance of Integrated Genetic Profiling in Adult T-Cell Acute Lymphoblastic Leukemia. Blood 2013, 122, 74–82.
- Vicente, C.; Schwab, C.; Broux, M.; Geerdens, E.; Degryse, S.; Demeyer, S.; Lahortiga, I.; Elliott, A.; Chilton, L.; La Starza, R.; et al. Targeted Sequencing Identifies Associations between IL7R-JAK Mutations and Epigenetic Modulators in T-Cell Acute Lymphoblastic Leukemia. Haematologica 2015, 100, 1301–1310.
- Genescà, E.; Lazarenkov, A.; Morgades, M.; Berbis, G.; Ruíz-Xivillé, N.; Gómez-Marzo, P.; Ribera, J.; Juncà, J.; González-Pérez, A.; Mercadal, S.; et al. Frequency and Clinical Impact of CDKN2A/ARF/CDKN2B Gene Deletions as Assessed by in-Depth Genetic Analyses in Adult T Cell Acute Lymphoblastic Leukemia. J. Hematol. Oncol. 2018, 11, 96.
- Van Vlierberghe, P.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Haydu, J.E.; Rigo, I.; Hadler, M.; Tosello, V.; Della Gatta, G.; Paietta, E.; Racevskis, J.; et al. ETV6 Mutations in Early Immature Human T Cell Leukemias. J. Exp. Med. 2011, 208, 2571–2579.
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The Genetic Basis of Early T-Cell Precursor Acute Lymphoblastic Leukaemia. Nature 2012, 481, 157–163.
- Neumann, M.; Heesch, S.; Schlee, C.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Konstandin, N.P.; Ksienzyk, B.; Vosberg, S.; Graf, A.; et al. Whole-Exome Sequencing in Adult ETP-ALL Reveals a High Rate of DNMT3A Mutations. Blood 2013, 121, 4749–4752.
- Bond, J.; Graux, C.; Lhermitte, L.; Lara, D.; Cluzeau, T.; Leguay, T.; Cieslak, A.; Trinquand, A.; Pastoret, C.; Belhocine, M.; et al. Early Response-Based Therapy Stratification Improves Survival in Adult Early Thymic Precursor Acute Lymphoblastic Leukemia: A Group for Research on Adult Acute Lymphoblastic Leukemia Study. J. Clin. Oncol. 2017, 35, 2683–2691.
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The Genomic Landscape of Pediatric and Young Adult T-Lineage Acute Lymphoblastic Leukemia. Nat. Genet. 2017, 49, 1211–1218.
- Gachet, S.; El-Chaar, T.; Avran, D.; Genesca, E.; Catez, F.; Quentin, S.; Delord, M.; Thérizols, G.; Briot, D.; Meunier, G.; et al. Deletion 6q Drives T-Cell Leukemia Progression by Ribosome Modulation. Cancer Discov. 2018, 8, 1614–1631.
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-Driven MYC Enhancer Promotes T Cell Development, Transformation and Acute Lymphoblastic Leukemia. Nat. Med. 2014, 20, 1130–1137.
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. Oncogene Regulation. An Oncogenic Super-Enhancer Formed through Somatic Mutation of a Noncoding Intergenic Element. Science 2014, 346, 1373–1377.
- Hu, S.; Qian, M.; Zhang, H.; Guo, Y.; Yang, J.; Zhao, X.; He, H.; Lu, J.; Pan, J.; Chang, M.; et al. Whole-Genome Noncoding Sequence Analysis in T-Cell Acute Lymphoblastic Leukemia Identifies Oncogene Enhancer Mutations. Blood 2017, 129, 3264–3268.
- Li, Z.; Abraham, B.J.; Berezovskaya, A.; Farah, N.; Liu, Y.; Leon, T.; Fielding, A.; Tan, S.H.; Sanda, T.; Weintraub, A.S.; et al. APOBEC Signature Mutation Generates an Oncogenic Enhancer That Drives LMO1 Expression in T-ALL. Leukemia 2017, 31, 2057–2064.
- Rahman, S.; Magnussen, M.; León, T.E.; Farah, N.; Li, Z.; Abraham, B.J.; Alapi, K.Z.; Mitchell, R.J.; Naughton, T.; Fielding, A.K.; et al. Activation of the LMO2 Oncogene through a Somatically Acquired Neomorphic Promoter in T-Cell Acute Lymphoblastic Leukemia. Blood 2017, 129, 3221–3226.
- Tottone, L.; Lancho, O.; Loh, J.-W.; Singh, A.; Kimura, S.; Roels, J.; Kuchmiy, A.; Strubbe, S.; Lawlor, M.A.; da Silva-Diz, V.; et al. A Tumor Suppressor Enhancer of PTEN in T-Cell Development and Leukemia. Blood Cancer Discov. 2021, 2, 92–109.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC Publications: Lyon, France, 2018; ISBN 978-92-832-4494-3.
- Morita, K.; Jain, N.; Kantarjian, H.; Takahashi, K.; Fang, H.; Konopleva, M.; El Hussein, S.; Wang, F.; Short, N.J.; Maiti, A.; et al. Outcome of T-Cell Acute Lymphoblastic Leukemia/Lymphoma: Focus on near-ETP Phenotype and Differential Impact of Nelarabine. Am. J. Hematol. 2021, 96, 589–598.
- Bond, J.; Touzart, A.; Leprêtre, S.; Graux, C.; Bargetzi, M.; Lhermitte, L.; Hypolite, G.; Leguay, T.; Hicheri, Y.; Guillerm, G.; et al. DNMT3A Mutation Is Associated with Increased Age and Adverse Outcome in Adult T-Cell Acute Lymphoblastic Leukemia. Haematologica 2019, 104, 1617–1625.
- González-Gil, C.; Morgades, M.; Fuster-Tormo, F.; García-Chica, J.; Montesinos, P.; Torrent, A.; Diaz-Beyá, M.; Coll, R.; Ribera, J.; Zhao, R.; et al. Genomic Data Improves Prognostic Stratification in Adult T-Cell Acute Lymphoblastic Leukemia Patients Enrolled in Measurable Residual Disease-Oriented Trials. Blood 2021, 138, 3486.
- Bond, J.; Marchand, T.; Touzart, A.; Cieslak, A.; Trinquand, A.; Sutton, L.; Radford-Weiss, I.; Lhermitte, L.; Spicuglia, S.; Dombret, H.; et al. An Early Thymic Precursor Phenotype Predicts Outcome Exclusively in HOXA-Overexpressing Adult T-Cell Acute Lymphoblastic Leukemia: A Group for Research in Adult Acute Lymphoblastic Leukemia Study. Haematologica 2016, 101, 732–740.
- Ben Abdelali, R.; Asnafi, V.; Petit, A.; Micol, J.-B.; Callens, C.; Villarese, P.; Delabesse, E.; Reman, O.; Lepretre, S.; Cahn, J.-Y.; et al. The Prognosis of CALM-AF10-Positive Adult T-Cell Acute Lymphoblastic Leukemias Depends on the Stage of Maturation Arrest. Haematologica 2013, 98, 1711–1717.
- Montefiori, L.E.; Bendig, S.; Gu, Z.; Chen, X.; Pölönen, P.; Ma, X.; Murison, A.; Zeng, A.; Garcia-Prat, L.; Dickerson, K.; et al. Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov. 2021, 11, 2846–2867.
- Di Giacomo, D.; La Starza, R.; Gorello, P.; Pellanera, F.; Kalender Atak, Z.; De Keersmaecker, K.; Pierini, V.; Harrison, C.J.; Arniani, S.; Moretti, M.; et al. 14q32 Rearrangements Deregulating BCL11B Mark a Distinct Subgroup of T-Lymphoid and Myeloid Immature Acute Leukemia. Blood 2021, 138, 773–784.
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The Genetic Basis and Cell of Origin of Mixed Phenotype Acute Leukaemia. Nature 2018, 562, 373–379.
- Takahashi, K.; Wang, F.; Morita, K.; Yan, Y.; Hu, P.; Zhao, P.; Zhar, A.A.; Wu, C.J.; Gumbs, C.; Little, L.; et al. Integrative Genomic Analysis of Adult Mixed Phenotype Acute Leukemia Delineates Lineage Associated Molecular Subtypes. Nat. Commun. 2018, 9, 2670.
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The Landscape of Somatic Mutations in Epigenetic Regulators across 1,000 Paediatric Cancer Genomes. Nat. Commun. 2014, 5, 3630.
- Christman, J.K. 5-Azacytidine and 5-Aza-2’-Deoxycytidine as Inhibitors of DNA Methylation: Mechanistic Studies and Their Implications for Cancer Therapy. Oncogene 2002, 21, 5483–5495.
- Batova, A.; Diccianni, M.B.; Yu, J.C.; Nobori, T.; Link, M.P.; Pullen, J.; Yu, A.L. Frequent and Selective Methylation of P15 and Deletion of Both P15 and P16 in T-Cell Acute Lymphoblastic Leukemia. Cancer Res. 1997, 57, 832–836.
- Tsellou, E.; Troungos, C.; Moschovi, M.; Athanasiadou-Piperopoulou, F.; Polychronopoulou, S.; Kosmidis, H.; Kalmanti, M.; Hatzakis, A.; Dessypris, N.; Kalofoutis, A.; et al. Hypermethylation of CpG Islands in the Promoter Region of the P15INK4B Gene in Childhood Acute Leukaemia. Eur. J. Cancer 2005, 41, 584–589.
- Takeuchi, S.; Matsushita, M.; Zimmermann, M.; Ikezoe, T.; Komatsu, N.; Seriu, T.; Schrappe, M.; Bartram, C.R.; Koeffler, H.P. Clinical Significance of Aberrant DNA Methylation in Childhood Acute Lymphoblastic Leukemia. Leuk. Res. 2011, 35, 1345–1349.
- Chim, C.S.; Tam, C.Y.; Liang, R.; Kwong, Y.L. Methylation of P15 and P16 Genes in Adult Acute Leukemia: Lack of Prognostic Significance. Cancer 2001, 91, 2222–2229.
- Garcia-Manero, G.; Bueso-Ramos, C.; Daniel, J.; Williamson, J.; Kantarjian, H.M.; Issa, J.-P.J. DNA Methylation Patterns at Relapse in Adult Acute Lymphocytic Leukemia. Clin. Cancer Res. 2002, 8, 1897–1903.
- Bueso-Ramos, C.; Xu, Y.; McDonnell, T.J.; Brisbay, S.; Pierce, S.; Kantarjian, H.; Rosner, G.; Garcia-Manero, G. Protein Expression of a Triad of Frequently Methylated Genes, P73, P57Kip2, and P15, Has Prognostic Value in Adult Acute Lymphocytic Leukemia Independently of Its Methylation Status. J. Clin. Oncol. 2005, 23, 3932–3939.
- Grossmann, V.; Haferlach, C.; Weissmann, S.; Roller, A.; Schindela, S.; Poetzinger, F.; Stadler, K.; Bellos, F.; Kern, W.; Haferlach, T.; et al. The Molecular Profile of Adult T-Cell Acute Lymphoblastic Leukemia: Mutations in RUNX1 and DNMT3A Are Associated with Poor Prognosis in T-ALL. Genes Chromosom. Cancer 2013, 52, 410–422.
- Jang, W.; Park, J.; Kwon, A.; Choi, H.; Kim, J.; Lee, G.D.; Han, E.; Jekarl, D.W.; Chae, H.; Han, K.; et al. CDKN2B Downregulation and Other Genetic Characteristics in T-Acute Lymphoblastic Leukemia. Exp. Mol. Med. 2019, 51, 1–15.
- Borssén, M.; Palmqvist, L.; Karrman, K.; Abrahamsson, J.; Behrendtz, M.; Heldrup, J.; Forestier, E.; Roos, G.; Degerman, S. Promoter DNA Methylation Pattern Identifies Prognostic Subgroups in Childhood T-Cell Acute Lymphoblastic Leukemia. PLoS ONE 2013, 8, e65373.
- Kimura, S.; Seki, M.; Kawai, T.; Goto, H.; Yoshida, K.; Isobe, T.; Sekiguchi, M.; Watanabe, K.; Kubota, Y.; Nannya, Y.; et al. DNA Methylation-Based Classification Reveals Difference between Pediatric T-Cell Acute Lymphoblastic Leukemia and Normal Thymocytes. Leukemia 2020, 34, 1163–1168.
- Touzart, A.; Boissel, N.; Belhocine, M.; Smith, C.; Graux, C.; Latiri, M.; Lhermitte, L.; Mathieu, E.-L.; Huguet, F.; Lamant, L.; et al. Low Level CpG Island Promoter Methylation Predicts a Poor Outcome in Adult T-Cell Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 1575–1581.
- Roels, J.; Thénoz, M.; Szarzyńska, B.; Landfors, M.; De Coninck, S.; Demoen, L.; Provez, L.; Kuchmiy, A.; Strubbe, S.; Reunes, L.; et al. Aging of Preleukemic Thymocytes Drives CpG Island Hypermethylation in T-Cell Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 274–289.
- Haider, Z.; Larsson, P.; Landfors, M.; Köhn, L.; Schmiegelow, K.; Flægstad, T.; Kanerva, J.; Heyman, M.; Hultdin, M.; Degerman, S. An Integrated Transcriptome Analysis in T-cell Acute Lymphoblastic Leukemia Links DNA Methylation Subgroups to Dysregulated TAL1 and ANTP Homeobox Gene Expression. Cancer Med. 2018, 8, 311–324.
- Maćkowska, N.; Drobna-Śledzińska, M.; Witt, M.; Dawidowska, M. DNA Methylation in T-Cell Acute Lymphoblastic Leukemia: In Search for Clinical and Biological Meaning. Int. J. Mol. Sci. 2021, 22, 1388.
More