Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 4 by Sirius Huang.
Gemcitabine is still the standard-of-care chemotherapeutic drug for pancreatic ductal adenocarcinoma (PDAC). However, the response rate is quite low. There are multiple mechanisms and participants in gemcitabine resistance.
- resistance to treatment
- gemcitabine
- desmoplastic reaction
- hydroxyurea
- proteasome inhibitors
1. Gemcitabine
Gemcitabine was introduced in pancreatic cancer treatment in 1997 after Burris et al. published their report [1]. This was a randomized clinical trial of 126 patients with advanced pancreatic cancer. They found that gemcitabine achieved better results than fluorouracil (5-FU) regarding a modest overall survival improvement and pain control. The mean survival only improved by one month (5.65 for gemcitabine vs. 4.41 for 5FU), but improvements in pain and the Karnofsky index were significant (23.8% for gemcitabine vs. 4.8% for 5FU).
Gemcitabine is still the standard-of-care chemotherapeutic drug for pancreatic ductal adenocarcinoma (PDAC) [2][3]. However, the response rate is quite low (around 30%), and even lower in advanced cases [4][5].
It improves average survival by two to three months [6], a really poor result. Chemoresistance develops rapidly [7] and is therefore the main limiting factor of the drug.
Gemcitabine is used as monotherapy or in combination with other chemotherapeutic drugs [8]. Results in combinatorial treatments are slightly better than monotherapy, however the high toxicity involved in combinatorial schemes led to its being used alone in many cases.
2. Mechanisms of Resistance to Gemcitabine
There are multiple mechanisms and participants in gemcitabine resistance. The following factors have been identified:
1. Decrease in deoxycytidine kinase expression or activity [9], thus impeding gemcitabine activation in this rate limiting step [10][11];
-
Decrease in deoxycytidine kinase expression or activity [9], thus impeding gemcitabine activation in this rate limiting step [10][11];
-
Increased expression of Ribonucleotide Reductase isoform M1 [12][13][14], leading to increased production of nucleotides for DNA synthesis;
-
c-Met activation: inhibition of c-Met with cabozantinib has overcome gemcitabine resistance and increased its cytotoxicity [20][21][22][23][24];
-
CAF-released exosomal miRNA 106-b [29]: CAFs are intrinsically resistant to gemcitabine and transmit this resistance through exosomes containing miRNA 106-b to cancer cells where they target TP53; For a review on miRNAs in pancreatic cancer, read Slotowinski et al. [30].
-
CAF production ofthe chemokine stromal cell-derived factor 1 (SDF1) is able to activate special AT-rich sequence-binding protein 1 (SATBP1), which intervenes in tumor progression and resistance to gemcitabine [31], as shown in the lower panel of Figure 1;
-
SDF-1α produced by stellate cells and secreted in the stroma has the ability to bind the CXCR4 over-expressed in pancreatic cancer cells, activating a pathway that increases survival, reduces apoptosis, and increases expression of IL-6 [32], as shown in Figure 1.
- Gemcitabine-resistant cells acquire EMT phenotype with cancer stem cell characteristics. Notch-2 and Jagged-1 are highly upregulated in these cells [36];
- Gemcitabine resistance-mediated EMT is in part induced by hypoxia because when HIF-1α is blocked, there is partial reversal of EMT [37];
- miR 233 is a contributing factor to gemcitabine resistance-dependent EMT [38];
- Cells that survive after gemcitabine treatment show increased stemness and EMT markers [39];
- Gemcitabine-induced EMT sustains chemoresistance [40];
- Targeting EMTcan overcome resistance [41].
- TGFβ1 also induces gemcitabine resistance
- [
- ]
- ;
- Epithelial–mesenchymal transition
- [
- ]
- , as the relationship between EMT and gemcitabine resistance is very complex:
-
Gemcitabine-resistant cells acquire EMT phenotype with cancer stem cell characteristics. Notch-2 and Jagged-1 are highly upregulated in these cells [36];
-
Gemcitabine resistance-mediated EMT is in part induced by hypoxia because when HIF-1α is blocked, there is partial reversal of EMT [37];
-
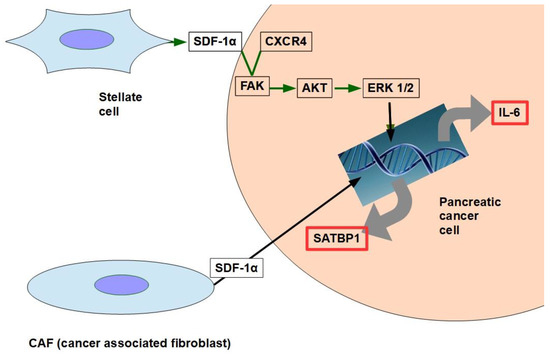
Figure 1. The two pathways shown in the figure have been found to decrease gemcitabine’s cytotoxicity and apoptosis. SDF-1α expression is induced by galectin 1.
Therefore, based on the above evidence, a circuit like the one shown below may represent the chain of events:
Gemcitabine→ EMT→ Resistance to Gemcitabine→ further EMT
- 16.
- 17.
- 18.
-
miRNA 320c through SMARCC1 (SMARCC1 is a protein that forms part of the SWI/SNF complex) [48]: This miRNA exerts contradictory actions because it has anti-tumor effects in bladder cancer by downregulating CDK6 [49] and in glioma, where it decreases growth and metastasis [50]. It was found to decrease canonical Wnt signaling in joints [51]. Therefore, miRNAwe 320c can be can considered miRNA 320can anti-oncogenic miRNA [52] which, however, promotes gemcitabine resistance.
- 19.
-
miRNA 21 and 221 [53][54]: miRNA 21 binds to the 3′-UTR region of the Bcl-2 gene, leading to its over-expression and thus inhibiting apoptosis of pancreatic cancer cells [55]; antisense miRNAs 21 and 221 restored gemcitabine sensitivity and induced cell-cycle arrest and apoptosis [53], while miRNA 200 [56][57] seems to antagonize miRNA21. miRNA 221 is considered a reliable circulating miRNA for diagnostic purposes [58];
- 20.
-
miRNA 155 modulates exosome synthesis and promotes gemcitabine resistance [59]; prolonged treatment with gemcitabine increased miRNA 155 levels, which in turn increased exosomes and expression of anti-apoptotic proteins. The message is carried to the rest of the cells through exosomes.
- 21.
-
miRNA 99a and miRNA100 [60] have been proposed as prognostic markers of gemcitabine resistance;
- 22.
- 23.
-
miRNA 365 induces gemcitabine resistance by targeting the anti-apoptotic BAX protein and its adaptor protein SHC1 [64]. It also induces the production of survival-related proteins;
- 24.
-
miRNA 210 [65] downregulates Homeobox protein Hox-A9, which increases NF-kB activity and decreases sensitivity to gemcitabine. However, the role of this microRNA is controversial. Amponsah et al. [66] identified miR-210 as a direct suppressor of the multidrug efflux transporter ABCC5; miR210 probably has dualpro- and anti-tumoral effects according to the balance with the oncomucin MUC4. They mutually regulate each other [67];
- 25.
The effects of some miRNAs regarding gemcitabine resistance are still a matter of debate. This is the case of miR-421, which seems to be pro-tumoral, reducing the expression of DPC4/SMAD4 [70] and at the same time increasing gemcitabine efficacy through decreased SPINK1 expression [71]. Furthermore, there is an oleic acid derivative, K73-3, that is able to upregulate miRNA 421 in vitro and in vivo, improving gemcitabine cytotoxicity [72]. Therefore, miRNA 421 should be considered an anti-oncogene agent.
In addition to the miRNAs discussed above, there are others without fully proven inhibitor effects on gemcitabine:
- 26.
-
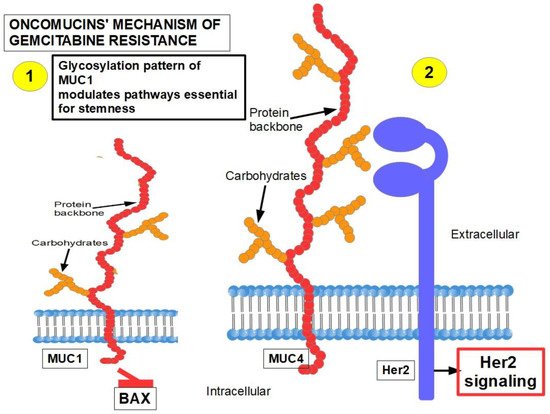
MUC1 and MUC4 [73][74] (Figure 2): Oncomucins play an important role in gemcitabine resistance that is discussed below. Mucins form a protective envelope surrounding cancer cells and participate in chemoresistance by impeding drug access to the malignant cells. Their production is usually highly increased in pancreatic cancer. There are two mechanisms involved in oncomucin-induced gemcitabine resistance:
-
- (1)
-
Direct, by MUC1 inhibiting the apoptotic BAX protein and increasing stemness;
-
- (2)
-
Indirect, by inducing Her 2 signaling.
-
-
-
Tumor-associated oncomucins have a different glycosylation pattern. MUC1 is less glycosylated than MUC4. MUC1-C, the intracellular portion of MUC1, is a driver for the upregulation of PD-L1. Although this immunoescape was found in triple-negative breast cancer, itwe can be hypothesized that pancreatic cancer’s refractoriness to immune-checkpoint inhibitors may be related to MUC1-C. MUC1-C expression also protects the malignant cells against genotoxic attacks in general;
MUC5AC, a facilitator of migration and invasion, also participates in drug resistance by inhibiting TRAIL death pathways.
- 27.
-
According to Shukla et al. [89], HIF-1α-dependent high glycolytic flux is the main player in gemcitabine resistance. High glycolytic flux allows for a high cytidine pool that competes with gemcitabine;
- 28.
-
CD44-expressing cells are resistant to gemcitabine. MDR1 is overexpressed in these cells [90]. These CSCs can rebuild the tumor after chemotherapy;
- 29.
-
Tumor heterogeneity: gemcitabine was more effective on cells that were more than 400–500 mμ from the desmoplastic areas [91]. Interestingly, high doses of metformin killed cells closer to the desmoplastic reaction area;
- 30.
-
ROCK2 (Rho associated protein kinase 2) activity is a cause of acquired gemcitabine resistance [92]. The Rhoa/ROCK2 axis promotes migration and metastasis. A pathway has been found in PDAC that shows the long non-coding RNA ZFAS1 inducing metastasis through the Rhoa/ROCK2 axis [93]. ZFAS1 is usually overexpressed in PDAC.ROCK inhibitors sensitize pancreatic CSCs to gemcitabine [94] and also reduce metastasis;
- 31.
-
Constitutive activation of NF-kB [95][96]: IL-1α expression is induced by NF-κB, which in turn increases NF-kB in a positive feedback loop, leading to permanent NF-kB activity [97][98][99][100]. In addition to the classic PI3K/AKT/NF-kB pathway that is fully operative in PDAC, several other pathways that induce gemcitabine resistance through NF-kB activity have been identified [101];
Pancreatic tumors show low miRNA 146-5p expression, impeding regulation of the TRAF6′s 3 UTR segment, thus allowing the pathway shown above. (TRAF6 is the tumor necrosis factor receptor-associated factor 6 that works as an adaptor protein allowing protein–protein interactions) [102].
PARP 14 (Poly ADP-ribose polymerase) is highly expressed in PDAC and is associated with poor prognosis. Silencing PARP 14 reduced resistance to gemcitabine.
Clusterin is a protein associated with chemoresistance to different chemotherapeutics. It was found to be increased in PDAC [103].
In summary, independently of which pathway activates NF-kB, this transcription factor has the ability to eliminate the pro-apoptotic effects of gemcitabine. Blocking NF-kB can, to a certain degree, decrease gemcitabine resistance [104].
- 32.
-
Increased expression of heme-oxygenase-1 (HO-1): PDAC cells show a 6-fold expression of HO-1 compared with normal pancreatic cells. Gemcitabine and/or radiotherapy treatments further increases HO-1 expression. HO-1 knockdown increases sensitivity to both therapies [105];
- 33.
-
Decreased expression of hENT1, the gemcitabine transporter, reduces its intracellular access [106];
- 34.
- 35.
-
Decreased glutathione peroxidase 1 induced resistance to gemcitabine: Glutathione peroxidase 1 modulates the AKT/GSK3β/Snail signaling axis in PDAC [110]. Interestingly, gemcitabine is able to induce the expression of glutathione pathway-related genes which are suspected of generating resistance [111];
- 36.
-
Increased expression of Snail [112];
- 37.
- 38.
-
Decreased intracellular ceramide/sphingosine-1-phosphate [115]: Increased ceramide favors apoptosis, while increased sphingosine-1-phosphate is anti-apoptotic; sphingosine kinase-1 is the enzyme that controls this ratio generating sphingosine-1-kinase, thus exerting anti-apoptotic effects;
- 39.
-
Mutation or deletion of the BRCA2 gene [116];
- 40.
-
Activation of Notch signaling [36][117][118] increases therapeutic resistance: This is related to the acquisition of an epithelial-mesenchymal phenotype (see paragraph 15); downregulation of Notch signaling has a chemosensitizing effect [119]; Notch-induced chemoresistance to gemcitabine is partly the result of Notch’s ability to alter the intrinsic apoptotic pathway [120];
- 41.
-
Hedgehog signaling [121]: chemotherapy activates the Hedgehog pathway [24], and this activation in turn leads to the expression of stem cell markers such as CD44, SOX2, OCT4, Nanog, and drug efflux proteins of the ATP-binding cassette family. Thus, Hedgehog increases stemness and induces a multidrug resistance phenotype [122];
- 42.
-
Cytosolic 5′-nucleotidase 1A over-expression [123]: This enzyme is able to reduce gemcitabine’s intracellular metabolites [124]. The histone deacetylase inhibitor trichostatin A has been found to synergize with gemcitabine, increasing its cytotoxicity, and importantly, inhibiting 5′-nucleotidase [125];
- 43.
-
Pancreatic cancer stem cells [126]: Stemness is a key factor in therapeutic failure in most tumors. CSCs do not respond to chemotherapy and are able to replicate the tumor after cytotoxic destruction of sensitive cells. The activation of pancreatic cancer stem cells has shown abilities to promote resistance to gemcitabine. Many of the activators are also involved in resistance. (Figure 3);
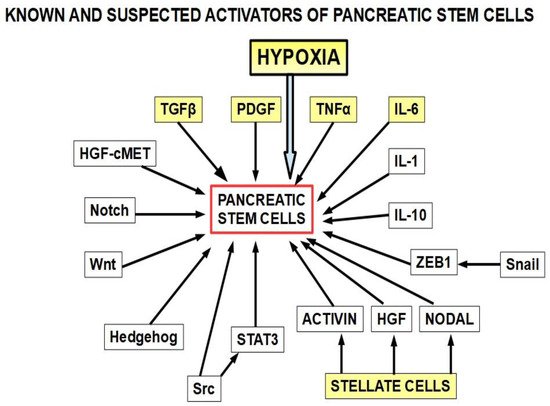 Figure 3. The yellow squares are the known activators of pancreatic cancer stem cells. The other activators (white squares) have also been found to play a role. This diagram is based on references [127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143][144][145][146]. Importantly, many of the stemness activators are also involved in epithelial–mesenchymal transition.
Figure 3. The yellow squares are the known activators of pancreatic cancer stem cells. The other activators (white squares) have also been found to play a role. This diagram is based on references [127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143][144][145][146]. Importantly, many of the stemness activators are also involved in epithelial–mesenchymal transition.
- 44.
- 45.
-
Calcyclin-binding protein or Siah-1-interacting protein (CacyBP/SIP) was found to be overexpressed in MDR after gemcitabine treatment. This protein induced P-gp and BCL2 expression reducing apoptosis [149]. In addition, CacyBP/SIP knockdown suppresses proliferation in pancreatic cancer by downregulating cyclin E and CDK2 and upregulating Rb and p27 [150];
- 46.
-
Soluble V-CAM, produced by pancreatic cancer cells, recruits tumor-associated macrophages (TAMs) [151];
- 47.
-
De novo lipid synthesis [152];
- 48.
-
The extracellular matrix composition: Laminin and collagen type IV-ECM (mimicking an early tumor ECM) protects from drug-induced apoptosis compared to a collagen I-rich late-tumor ECM;
- 49.
- 50.
-
Autophagy has been shown to be upregulated in PDAC and it plays an important role in resistance to chemotherapy [158][159]. Autophagy is an inducer of gemcitabine resistance and is probably one of the mechanisms that cells use to survive cytotoxic drugs. Gemcitabine’s cytotoxicity is increased when an autophagy-inhibitor is used simultaneously [160]. Pancreatic adenocarcinoma is a very hypoxic tumor, and hypoxia can induce autophagy. Additionally, the expression of high-mobility group box 1 (HMGB1) is an autophagy inducer. Interestingly, gemcitabine upregulates this protein, thus indirectly increasing autophagy [161]. In a preoperative setting, when combining the autophagy inhibitor hydroxychloroquine with gemcitabine, 61% of patients showed CA19.9 marker decrease, improved postoperative, and disease-free survival. These findings were particularly evident in the patients with high levels of the autophagy marker LC3-II [162]. By blocking autophagy, gemcitabine’s cytotoxic effects were increased and stem cell activity reduced [163]. Zeh et al. [164] studied two cohorts of preoperative patients, one receiving nab-placlitaxel, and another group with the same medication plus hydroxychloroquine. They found that the resected pancreas in the hydroxychloroquine group had a greater pathologic response and higher immune activity. However, overall survival and disease-free survival was similar in both cohorts. SNHG14 (small nuclear RNA host gene 14) oncogene expression generates a long non-coding RNA that induces autophagy and resistance to gemcitabine [165]. This LNC-RNA seems to act as an anti-sense against MiRs involved in anti-tumoral activity;
The conclusion is that there is evidence supporting better results with longer overall survival and disease-free survival by adding autophagy inhibitors to gemcitabine in the resectable cases [166]. Evidence in this regard is lacking for inoperable patients.
- 51.
-
Pancreatic tumor microbiota: There is a clinically important population of bacteria and fungi within the pancreas and biliary tree in patients with PDAC and this population is different from the microbiota found in the normal pancreas [167][168][169]. The bacteria present in PDAC show some specificity [170]. Regarding gemcitabine, it was found that intratumoral Gammaproteobacteriahad a role in resistance [171]. Patients that had some surgical or endoscopic procedure on the pancreas and the biliary tree were prone to host pro-resistance bacteria in the pancreas [172], and 5-FU resistance was associated with the presence of Fusobacterium nucleatum in colorectal cancer [173]. Fusobacterium is very abundant in PDAC, so it can be hypothesized that it also plays a role in pancreatic chemoresistance. Furthermore, Fusobacterium induces autophagy as part of its chemoresistance mechanism, another frequent finding in PDAC. Fungi have also been found to be a possible cause of gemcitabine resistance [174];
- 52.
-
Hypoxia is a key factor in the PDAC phenotype, including proliferation, autophagy, progression, metastasis, as well as resistance to treatment in general, and to gemcitabine in particular [175]. The evidence is compelling [176][177][178][179][180][181][182][183]. A simple example shows the importance of this issue. Hypoxia is expressed through the hypoxia-inducible factors (transcription factors that modulate over 150 genes). Downregulation of HIF-1α with a newly developed molecule, LW6, inhibited autophagic flux, improved the efficacy of gemcitabine, stopped proliferation, and induced cell death [184]. LW6 is a novel HIF-1 inhibitor that decreases HIF-1α protein expression [185][186][187];
Hypoxia not only increases resistance to gemcitabine, it also increases gemcitabine-induced stemness [188]. Luo et al. [189] showed that hypoxia-induced miRNA 301a which in turn promoted gemcitabine resistance through downregulation of T53, thereby integrating hypoxia, miR, and gemcitabine resistance into one pathway.

Figure 4. Mechanism of hypoxia-induced gemcitabine resistance.
- 53.
-
Increased expression of cytoplasmic ribonucleotide reductase subunit M1 (RRM1) [190]: RR is a multimeric enzyme essential for maintaining a high pool of deoxynucleotides for DNA elongation and also for DNA repair. Gemcitabine-resistant pancreatic cancer cells treated with RRM1 inhibitors showed considerable decrease in resistance [13]. Patients with high RRM1 levels showed a poorer overall survival with gemcitabine treatment compared with low RRM1- expressing patients [191];
- 54.
Figure 5 presents a summary of mechanisms involved in gemcitabine resistance.
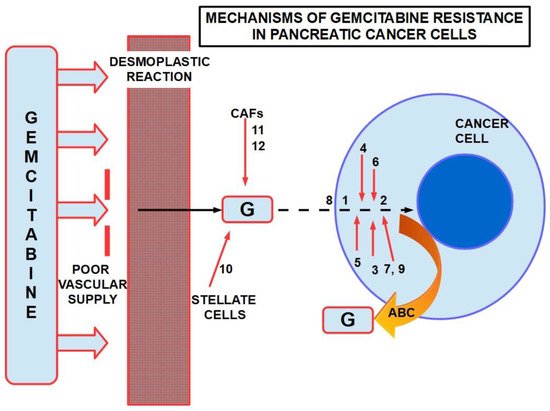
Figure 5. Some mechanisms of resistance to gemcitabine in PDAC. ABC: ATP binding cassette. Poor vascular supply and the desmoplastic reactionare mainly physical barriers. The numbers are chemicals and pathways activated for the escape. ABC re-exports the cytotoxic substances.
References
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413.
- Manji, G.A.; Olive, K.; Saenger, Y.M.; Oberstein, P. Current and emerging therapies in metastatic pancreatic cancer. Clin. Cancer Res. 2017, 23, 1670–1678.
- Conroy, T.; Bachet, J.-B.; Ayav, A.; Huguet, F.; Lambert, A.; Caramella, C.; Maréchal, R.; Van Laethem, J.-L.; Ducreux, M. Current standards and new innovative approaches for treatment of pancreatic cancer. Eur. J. Cancer 2016, 57, 10–22.
- Hashimoto, K.; Ueno, H.; Ikeda, M.; Kojima, Y.; Hagihara, A.; Kondo, S.; Morizane, C.; Okusaka, T. Do recurrent and metastatic pancreatic cancer patients have the same outcomes with gemcitabine treatment? Oncology 2009, 77, 217–223.
- Andriulli, A.; Festa, V.; Botteri, E.; Valvano, M.R.; Koch, M.; Bassi, C.; Maisonneuve, P.; Di Sebastiano, P. Neoadjuvant/preoperative gemcitabine for patients with localized pancreatic cancer: A meta-analysis of prospective studies. Ann. Surg. Oncol. 2011, 19, 1644–1662.
- Ahmed, A.A.; Marchetti, C.; Ohnmacht, S.A.; Neidle, S. A G-quadruplex-binding compound shows potent activity in human gemcitabine-resistant pancreatic cancer cells. Sci. Rep. 2020, 10, 1–11.
- Amrutkar, M.; Gladhaug, I.P. Pancreatic cancer chemoresistance to gemcitabine. Cancers 2017, 9, 157.
- Zhang, X.-W.; Ma, Y.-X.; Sun, Y.; Cao, Y.-B.; Li, Q.; Xu, C.-A. Gemcitabine in combination with a second cytotoxic agent in the first-line treatment of locally advanced or metastatic pancreatic cancer: A systematic review and meta-analysis. Target. Oncol. 2017, 65, 5–321.
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285.
- Sebastiani, V.; Ricci, F.; Rubio-Viquiera, B.; Kulesza, P.; Yeo, C.J.; Hidalgo, M.; Klein, A.; Laheru, D.; Iacobuzio-Donahue, C.A. Immunohistochemical and genetic evaluation of deoxycytidine kinase in pancreatic cancer: Relationship to molecular mechanisms of gemcitabine resistance and survival. Clin. Cancer Res. 2006, 12, 2492–2497.
- de Sousa Cavalcante, L.; Monteiro, G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014, 741, 8–16.
- Bergman, A.M.; Eijk, P.P.; van Haperen, V.W.R.; Smid, K.; Veerman, G.; Hubeek, I.; Ijssel, P.V.D.; Ylstra, B.; Peters, G.J. In Vivo induction of resistance to gemcitabine results in increased expression of ribonucleotide reductase subunit m1 as the major determinant. Cancer Res. 2005, 65, 9510–9516.
- Nakahira, S.; Nakamori, S.; Tsujie, M.; Takahashi, Y.; Okami, J.; Yoshioka, S.; Yamasaki, M.; Marubashi, S.; Takemasa, I.; Miyamoto, A.; et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int. J. Cancer 2006, 120, 1355–1363.
- Akita, H.; Zheng, Z.; Takeda, Y.; Kim, C.; Kittaka, N.; Kobayashi, S.; Marubashi, S.; Takemasa, I.; Nagano, H.; Dono, K.; et al. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene 2009, 28, 2903–2909.
- Bondar, V.M.; Sweeney-Gotsch, B.; Andreeff, M.; Mills, G.B.; McConkey, D.J. Inhibition of the phosphatidyl-inositol 3′-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and In Vivo. Mol. Cancer Ther. 2002, 1, 989–997.
- Erkan, M.; Kleeff, J.; Esposito, I.; Giese, T.; Ketterer, K.; Büchler, M.W.; A Giese, N.; Friess, H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 2005, 24, 4421–4432.
- Duxbury, M.; Ito, H.; Zinner, M.; Ashley, S.; Whang, E. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery 2004, 135, 555–562.
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; E Whang, E. siRNA directed against c-Src enhances pancreatic adenocarcinoma cell gemcitabine chemosensitivity. J. Am. Coll. Surg. 2004, 198, 953–959.
- Shah, A.N.; Gallick, G.E. Src, chemoresistance and epithelial to mesenchymal transition: Are they related? Anti-Cancer Drugs 2007, 18, 371–375.
- Hage, C.; Rausch, V.; Giese, N.; Giese, T.; Schönsiegel, F.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Herr, I. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Cell Death Dis. 2013, 4, e627.
- Noguchi, K.; Konno, M.; Eguchi, H.; Kawamoto, K.; Mukai, R.; Nishida, N.; Koseki, J.; Wada, H.; Akita, H.; Satoh, T.; et al. c-Met affects gemcitabine resistance during carcinogenesis in a mouse model of pancreatic cancer. Oncol. Lett. 2018, 16, 1892–1898.
- Brandes, F.; Schmidt, K.; Wagner, C.; Redekopf, J.; Schlitt, H.J.; Geissler, E.K.; Lang, S.A. Targeting cMET with INC280 impairs tumour growth and improves efficacy of gemcitabine in a pancreatic cancer model. BMC Cancer 2015, 15, 1–14.
- Avan, A.; Quint, K.; Nicolini, F.; Funel, N.; Frampton, A.E.; Maftouh, M.; Pelliccioni, S.; Schuurhuis, G.J.; Peters, G.J.; Giovannetti, E. Enhancement of the antiproliferative activity of gemcitabine by modulation of c-Met pathway in pancreatic cancer. Curr. Pharm. Des. 2013, 19, 940–950.
- Firuzi, O.; Che, P.P.; El Hassouni, B.; Buijs, M.; Coppola, S.; Löhr, M.; Funel, N.; Heuchel, R.; Carnevale, I.; Schmidt, T.; et al. Role of c-MET inhibitors in overcoming drug resistance in spheroid models of primary human pancreatic cancer and stellate cells. Cancers 2019, 11, 638.
- Liau, S.; Ashley, S.; Whang, E. Lentivirus-mediated RNA interference of HMGA1 promotes chemosensitivity to gemcitabine in pancreatic adenocarcinoma. J. Gastrointest. Surg. 2006, 10, 1254–1263.
- Liau, S.-S.; Whang, E. HMGA1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin. Cancer Res. 2008, 14, 1470–1477.
- Liau, S.-S.; Jazag, A.; Whang, E.E. HMGA1 is a determinant of cellular invasiveness and In Vivo metastatic potential in pancreatic adenocarcinoma. Cancer Res. 2006, 66, 11613–11622.
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Heiden, M.G.V.; et al. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res. 2019, 79, 5723–5733.
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543.
- Słotwiński, R.; Lech, G.; Słotwińska, S.M. MicroRNAs in pancreatic cancer diagnosis and therapy. Cent. Eur. J. Immunol. 2018, 43, 314–324.
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065.
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine SDF-1α signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2014, 6, 3085–3097.
- Yu, C.; Chen, S.; Guo, Y.; Sun, C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 2018, 8, 3224–3236.
- Zhuang, J.; Shen, L.; Yang, L.; Huang, X.; Lu, Q.; Cui, Y.; Zheng, X.; Zhao, X.; Zhang, D.; Huang, R.; et al. TGFβ1 promotes gemcitabine resistance through regulating the LncRNA-LET/NF90/miR-145 signaling axis in bladder cancer. Theranostics 2017, 7, 3053–3067.
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial—Mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352.
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407.
- Wang, R.; Cheng, L.; Xia, J.; Wang, Z.; Wu, Q.; Wang, Z. Gemcitabine resistance is associated with epithelial-mesenchymal transition and induction of HIF-1α in pancreatic cancer cells. Curr. Cancer Drug Targets 2014, 14, 407–417.
- Ma, J.; Fang, B.; Zeng, F.; Ma, C.; Pang, H.; Cheng, L.; Shi, Y.; Wang, H.; Yin, B.; Xia, J.; et al. Down-regulation of miR-223 reverses epithelial-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Oncotarget 2015, 6, 1740–1749.
- Quint, K.; Tonigold, M.; Di Fazio, P.; Montalbano, R.; Lingelbach, S.; Rückert, F.; Alinger, B.; Ocker, M.; Neureiter, D. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. Int. J. Oncol. 2012, 41, 2093–2102.
- El Amrani, M.; Corfiotti, F.; Corvaisier, M.; Vasseur, R.; Fulbert, M.; Skrzypczyk, C.; Deshorgues, A.; Gnemmi, V.; Tulasne, D.; Lahdaoui, F.; et al. Gemcitabine-induced epithelial-mesenchymal transition-like changes sustain chemoresistance of pancreatic cancer cells of mesenchymal-like phenotype. Mol. Carcinog. 2019, 58, 1985–1997.
- Elaskalani, O.; Razak, N.B.A.; Falasca, M.; Metharom, P. Epithelial-mesenchymal transition as a therapeutic target for overcoming chemoresistance in pancreatic cancer. World J. Gastrointest. Oncol. 2017, 9, 37–41.
- Adamska, A.; Falasca, M. ATP-binding cassette transporters in progression and clinical outcome of pancreatic cancer: What is the way forward? World J. Gastroenterol. 2018, 24, 3222–3238.
- Sasaki, N.; Ishiwata, T.; Hasegawa, F.; Michishita, M.; Kawai, H.; Matsuda, Y.; Arai, T.; Ishikawa, N.; Aida, J.; Takubo, K.; et al. Stemness and anti-cancer drug resistance in ATP-binding cassette subfamily G member 2 highly expressed pancreatic cancer is induced in 3D culture conditions. Cancer Sci. 2018, 109, 1135–1146.
- Mohelnikova-Duchonova, B.; Brynychova, V.; Oliverius, M.; Hlavsa, J.; Honsova, E.; Mazanec, J.; Melichar, B.; Kala, Z.; Soucek, P. Prognostic significance of ATP-binding cassette (ABC) and solute carrier (SLC) transporters in pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2013, 31, 11051.
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12–CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679.
- Aravin, A.A.; Lagos-Quintana, M.; Yalcin, A.; Zavolan, M.; Marks, D.; Snyder, B.; Gaasterland, T.; Meyer, J.; Tuschl, T. The small RNA profile during drosophila melanogaster development. Dev. Cell 2003, 5, 337–350.
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297.
- Iwagami, Y.; Eguchi, H.; Nagano, H.; Akita, H.; Hama, N.; Wada, H.; Kawamoto, K.; Kobayashi, S.; Tomokuni, A.; Tomimaru, Y.; et al. Mir-320c regulates gemcitabine-resistance in pancreatic cancer via SMARCC1. Br. J. Cancer 2013, 109, 502–511.
- Wang, X.; Wu, J.; Lin, Y.; Zhu, Y.; Xu, X.; Xu, X.; Liang, Z.; Li, S.; Hu, Z.; Zheng, X.; et al. MicroRNA-320c inhibits tumorous behaviors of bladder cancer by targeting cyclin-dependent kinase 6. J. Exp. Clin. Cancer Res. 2014, 33, 69.
- Lv, Q.-L.; Zhu, H.-T.; Li, H.-M.; Cheng, X.-H.; Zhou, H.-H.; Chen, S.-H. Down-regulation of miRNA-320c promotes tumor growth and metastasis and predicts poor prognosis in human glioma. Brain Res. Bull. 2018, 139, 125–132.
- Hu, S.; Mao, G.; Zhang, Z.; Wu, P.; Wen, X.; Liao, W.; Zhang, Z. MicroRNA-320c inhibits development of osteoarthritis through downregulation of canonical Wnt signaling pathway. Life Sci. 2019, 228, 242–250.
- Liang, Y.; Li, S.; Tang, L. MicroRNA 320, an anti-oncogene target miRNA for cancer therapy. Biomedicines 2021, 9, 591.
- Park, J.-K.; Lee, E.J.; Esau, C.; Schmittgen, T.D. Antisense Inhibition of microRNA-21 or -221 Arrests Cell Cycle, induces apoptosis, and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma. Pancreas 2009, 38, e190–e199.
- Giovannetti, E.; Funel, N.; Peters, G.J.; Del Chiaro, M.; Erozenci, L.A.; Vasile, E.; Leon, L.G.; Pollina, L.E.; Groen, A.; Falcone, A.; et al. MicroRNA-21 in pancreatic cancer: Correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res. 2010, 70, 4528–4538.
- Dong, J.; Zhao, Y.-P.; Zhou, L.; Zhang, T.-P.; Chen, G. Bcl-2 upregulation induced by miR-21 via a direct interaction is associated with apoptosis and chemoresistance in MIA PaCa-2 pancreatic cancer cells. Arch. Med Res. 2011, 42, 8–14.
- Ali, S.; Ahmad, A.; Banerjee, S.; Padhye, S.; Dominiak, K.; Schaffert, J.M.; Wang, Z.; Philip, P.A.; Sarkar, F.H. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010, 70, 3606–3617.
- Li, Y.; Vandenboom, T.G., II; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712.
- Kawaguchi, T.; Komatsu, S.; Ichikawa, D.; Morimura, R.; Tsujiura, M.; Konishi, H.; Takeshita, H.; Nagata, H.; Arita, T.; Hirajima, S.; et al. Clinical impact of circulating miR-221 in plasma of patients with pancreatic cancer. Br. J. Cancer 2013, 108, 361–369.
- Mikamori, M.; Yamada, D.; Eguchi, H.; Hasegawa, S.; Kishimoto, T.; Tomimaru, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; et al. MicroRNA-155 controls exosome synthesis and promotes gemcitabine resistance in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 42339.
- Dhayat, S.A.; Abdeen, B.; Köhler, G.; Senninger, N.; Haier, J.; Mardin, W.A. MicroRNA-100 and microRNA-21 as markers of survival and chemotherapy response in pancreatic ductal adenocarcinoma UICC stage II. Clin. Epigenetics 2015, 7, 1–15.
- Zhang, X.J.; Ye, H.; Zeng, C.W.; He, B.; Zhang, H.; Chen, Y.Q. Dysregulation of miR-15a and miR-214 in human pancreatic cancer. J. Hematol. Oncol. 2010, 3, 46.
- Kuninty, P.R.; Bojmar, L.; Tjomsland, V.; Larsson, M.; Storm, G.; Östman, A.; Sandström, P.; Prakash, J. MicroRNA-199a and -214 as potential therapeutic targets in pancreatic stellate cells in pancreatic tumor. Oncotarget 2016, 7, 16396–16408.
- Wang, Y.-S.; Tian, J.; Han, Y.; Han, S.-M.; Shi, S.-B. Gemcitabine plus vinorelbine as second-line therapy in patients with metastatic esophageal cancer previously treated with platinum-based chemotherapy. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2016, 24, 129–135.
- Hamada, S.; Masamune, A.; Miura, S.; Satoh, K.; Shimosegawa, T. MiR-365 induces gemcitabine resistance in pancreatic cancer cells by targeting the adaptor protein SHC1 and pro-apoptotic regulator BAX. Cell. Signal. 2013, 26, 179–185.
- Ni, J.; Zhou, S.; Yuan, W.; Cen, F.; Yan, Q. Mechanism of miR-210 involved in epithelial—Mesenchymal transition of pancreatic cancer cells under hypoxia. J. Recept. Signal Transduct. 2019, 39, 399–406.
- Amponsah, P.S.; Fan, P.; Bauer, N.; Zhao, Z.; Gladkich, J.; Fellenberg, J.; Herr, I. MicroRNA-210 overexpression inhibits tumor growth and potentially reverses gemcitabine resistance in pancreatic cancer. Cancer Lett. 2017, 388, 107–117.
- Boukrout, N.; Souidi, M.; Lahdaoui, F.; Duchêne, B.; Neve, B.; Coppin, L.; Leteurtre, E.; Torrisani, J.; Van Seuningen, I.; Jonckheere, N. Antagonistic roles of the tumor suppressor miR-210-3p and oncomucin MUC4 forming a negative feedback loop in pancreatic adenocarcinoma. Cancers 2021, 13, 6197.
- Yu, J.; Ohuchida, K.; Mizumoto, K.; Fujita, H.; Nakata, K.; Tanaka, M. MicroRNAmiR-17-5pis overexpressed in pancreatic cancer, associated with a poor prognosis, and involved in cancer cell proliferation and invasion. Cancer Biol. Ther. 2010, 10, 748–757.
- Yan, H.-J.; Liu, W.-S.; Sun, W.-H.; Wu, J.; Ji, M.; Wang, Q.; Zheng, X.; Jiang, J.-T.; Wu, C.-P. MiR-17-5p inhibitor enhances chemosensitivity to gemcitabine via upregulating bim expression in pancreatic cancer cells. Am. J. Dig. Dis. 2012, 57, 3160–3167.
- Hao, J.; Zhang, S.; Zhou, Y.; Liu, C.; Hu, X.; Shao, C. MicroRNA 421 suppresses DPC4/Smad4 in pancreatic cancer. Biochem. Biophys. Res. Commun. 2011, 406, 552–557.
- Shopit, A.; Li, X.; Wang, S.; Awsh, M.; Safi, M.; Chu, P.; Jia, J.; Al-Radhi, M.; Baldi, S.; Wang, F.; et al. Enhancement of gemcitabine efficacy by K73-03 via epigenetically regulation of miR-421/SPINK1 in gemcitabine resistant pancreatic cancer cells. Phytomedicine 2021, 91, 153711.
- Shopit, A.; Li, X.; Tang, Z.; Awsh, M.; Shobet, L.; Niu, M.; Wang, H.; Mousa, H.; Alshwmi, M.; Tesfaldet, T.; et al. MiR-421 up-regulation by the oleanolic acid derivative K73-03 regulates epigenetically SPINK1 transcription in pancreatic cancer cells leading to metabolic changes and enhanced apoptosis. Pharmacol. Res. 2020, 161, 105130.
- Rachagani, S.; Macha, M.; Ponnusamy, M.P.; Haridas, D.; Kaur, S.; Jain, M.; Batra, S.K. MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis 2012, 33, 1953–1964.
- Torres, M.P.; Chakraborty, S.; Souchek, J.; Batra, S.K. Mucin-based targeted pancreatic cancer therapy. Curr. Pharm. Des. 2012, 18, 2472–2481.
- Barkeer, S.; Chugh, S.; Batra, S.K.; Ponnusamy, M.P. Glycosylation of cancer stem cells: Function in stemness, tumorigenesis, and metastasis. Neoplasia 2018, 20, 813–825.
- Hossain, K.; Wall, K.A. Immunological evaluation of recent MUC1 glycopeptide cancer vaccines. Vaccines 2016, 4, 25.
- Beckwith, D.M.; Cudic, M. Tumor-associated O-glycans of MUC1: Carriers of the glyco-code and targets for cancer vaccine design. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2020; Volume 47, p. 101389.
- Li, W.; Zhang, N.; Jin, C.; Long, M.D.; Rajabi, H.; Yasumizu, Y.; Fushimi, A.; Yamashita, N.; Hagiwara, M.; Zheng, R.; et al. MUC1-C drives stemness in progression of colitis to colorectal cancer. JCI Insight 2020, 5, e137112.
- Huang, W.-C.; Chan, M.-L.; Chen, M.-J.; Tsai, T.-H.; Chen, Y.-J. Modulation of macrophage polarization and lung cancer cell stemness by MUC1 and development of a related small-molecule inhibitor pterostilbene. Oncotarget 2016, 7, 39363–39375.
- Rogoff, H.A.; Li, J.; Li, C. Abstract 4777: Cancer stemness and resistance: Napabucasin (BBI-608) sensitizes stemness-high cancer cells to paclitaxel by inhibiting the STAT3-MUC1 pathway. Cancer Res. 2017, 77, 4777.
- Hata, T.; Rajabi, H.; Yamamoto, M.; Jin, C.; Ahmad, R.; Zhang, Y.; Kui, L.; Li, W.; Yasumizu, Y.; Hong, D.; et al. Targeting MUC1-C inhibits TWIST1 signaling in triple-negative breast cancer. Mol. Cancer Ther. 2019, 18, 1744–1754.
- Ham, S.Y.; Kwon, T.; Bak, Y.; Yu, J.-H.; Hong, J.; Lee, S.K.; Yu, D.-Y.; Yoon, D.-Y. Mucin 1-mediated chemo-resistance in lung cancer cells. Oncogenesis 2016, 5, e185.
- Alam, M.; Ahmad, R.; Rajabi, H.; Kufe, D. MUC1-C induces the LIN28B→LET-7→HMGA2 axis to regulate self-renewal in NSCLC. Mol. Cancer Res. 2015, 13, 449–460.
- Ahmad, R.; Alam, M.; Rajabi, H.; Kufe, D. The MUC1-C oncoprotein binds to the BH3 domain of the pro-apoptotic BAX protein and blocks BAX function. J. Biol. Chem. 2012, 287, 20866–20875.
- Maeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C induces PD-L1 and immune evasion in triple-negative breast cancer. Cancer Res. 2018, 78, 205–215.
- Ren, J.; Agata, N.; Chen, D.; Li, Y.; Yu, W.-H.; Huang, L.; Raina, D.; Chen, W.; Kharbanda, S.; Kufe, D. Human MUC1 carcinoma-associated protein confers resistance to genotoxic anticancer agents. Cancer Cell 2004, 5, 163–175.
- Jonckheere, N.; Skrypek, N.; Merlin, J.; Dessein, A.F.; Dumont, P.; Leteurtre, E.; Harris, A.; Desseyn, J.-L.; Susini, C.; Frénois, F.; et al. The mucin MUC4 and its membrane partner ErbB2 regulate biological properties of human CAPAN-2 pancreatic cancer cells via different signalling pathways. PLoS ONE 2012, 7, e32232.
- Skrypek, N.; Vasseur, R.; Vincent, A.; Duchene, B.; Van Seuningen, I.; Jonckheere, N. The oncogenic receptor ErbB2 modulates gemcitabine and irinotecan/SN-38 chemoresistance of human pancreatic cancer cells via hCNT1 transporter and multidrug-resistance associated protein MRP-2. Oncotarget 2015, 6, 10853–10867.
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 2017, 32, 71–87.
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331.
- Zechner, D.; Bürtin, F.; Albert, A.-C.; Zhang, X.; Kumstel, S.; Schönrogge, M.; Graffunder, J.; Shih, H.-Y.; Müller, S.; Radecke, T.; et al. Intratumoral heterogeneity of the therapeutical response to gemcitabine and metformin. Oncotarget 2016, 7, 56395–56407.
- Zhou, Y.; Zhou, Y.; Wang, K.; Li, T.; Zhang, M.; Yang, Y.; Wang, R.; Hu, R. ROCK2 confers acquired gemcitabine resistance in pancreatic cancer cells by upregulating transcription factor ZEB1. Cancers 2019, 11, 1881.
- Liu, J.; Zhu, Y.; Ge, C. LncRNA ZFAS1 promotes pancreatic adenocarcinoma metastasis via the RHOA/ROCK2 pathway by sponging miR-3924. Cancer Cell Int. 2020, 20, 1–15.
- Takeda, H.; Okada, M.; Suzuki, S.; Kuramoto, K.; Sakaki, H.; Watarai, H.; Sanomachi, T.; Seino, S.; Yoshioka, T.; Kitanaka, C. Rho-associated protein kinase (ROCK) inhibitors inhibit survivin expression and sensitize pancreatic cancer stem cells to gemcitabine. Anticancer Res. 2016, 36, 6311–6318.
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127.
- Weichert, W.; Boehm, M.F.; Gekeler, V.; Bahra, M.; Langrehr, J.M.; Neuhaus, P.; Denkert, C.; Imre, G.; Weller, C.M.; Hofmann, H.-P.; et al. High expression of RelA/p65 is associated with activation of nuclear factor-κB-dependent signaling in pancreatic cancer and marks a patient population with poor prognosis. Br. J. Cancer 2007, 97, 523–530.
- Melisi, D.; Niu, J.; Chang, Z.; Xia, Q.; Peng, B.; Ishiyama, S.; Evans, D.B.; Chiao, P.J. Secreted interleukin-1α induces a metastatic phenotype in pancreatic cancer by sustaining a constitutive activation of nuclear factor-κB. Mol. Cancer Res. 2009, 7, 624–633.
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120.
- Arlt, A.; Vorndamm, J.; Breitenbroich, M.; Fölsch, U.R.; Kalthoff, H.; E Schmidt, W.; Schäfer, H. Inhibition of NF-κB sensitizes human pancreatic carcinoma cells to apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene 2001, 20, 859–868.
- Arlt, A.; Gehrz, A.; Müerköster, S.; Vorndamm, J.; Kruse, M.-L.; Fölsch, U.R.; Schäfer, H. Role of NF-κB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003, 22, 3243–3251.
- Meng, Q.; Liang, C.; Hua, J.; Zhang, B.; Liu, J.; Zhang, Y.; Wei, M.; Yu, X.; Xu, J.; Shi, S. A miR-146a-5p/TRAF6/NF-kB p65 axis regulates pancreatic cancer chemoresistance: Functional validation and clinical significance. Theranostics 2020, 10, 3967–3979.
- Yao, N.; Chen, Q.; Shi, W.; Tang, L.; Fu, Y. PARP14 promotes the proliferation and gemcitabine chemoresistance of pancreatic cancer cells through activation of NF-κB pathway. Mol. Carcinog. 2019, 58, 1291–1302.
- Xu, M.; Chen, X.; Han, Y.; Ma, C.; Ma, L.; Li, S. Clusterin silencing sensitizes pancreatic cancer MIA-PaCa-2 cells to gmcitabine via regulation of NF-kB/Bcl-2 signaling. Int. J. Clin. Exp. Med. 2015, 8, 12476.
- Li, Q.; Yang, G.; Feng, M.; Zheng, S.; Cao, Z.; Qiu, J.; You, L.; Zheng, L.; Hu, Y.; Zhang, T.; et al. NF-κB in pancreatic cancer: Its key role in chemoresistance. Cancer Lett. 2018, 421, 127–134.
- Berberat, P.O.; Dambrauskas, Z.; Gulbinas, A.; Giese, T.; Giese, N.; Künzli, B.; Autschbach, F.; Meuer, S.; Büchler, M.W.; Friess, H. Inhibition of heme Oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 2005, 11, 3790–3798.
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; A Belt, J.; Crawford, C.R.; E Cass, C. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357.
- Yu, C.; Zhang, X.; Sun, G.; Guo, X.; Li, H.; You, Y.; Jacobs, J.L.; Gardner, K.; Yuan, D.; Xu, Z.; et al. RNA interference-mediated silencing of thepolo-like kinase 1gene enhances chemosensitivity to gemcitabine in pancreatic adenocarcinoma cells. J. Cell. Mol. Med. 2008, 12, 2334–2349.
- Mao, Y.; Xi, L.; Li, Q.; Cai, Z.; Lai, Y.; Zhang, X.; Yu, C. Regulation of cell apoptosis and proliferation in pancreatic cancer through PI3K/Akt pathway via Polo-like kinase 1. Oncol. Rep. 2016, 36, 49–56.
- Ma, W.W.; Messersmith, W.A.; Dy, G.K.; Weekes, C.D.; Whitworth, A.; Ren, C.; Maniar, M.; Wilhelm, F.; Eckhardt, S.G.; Adjei, A.A.; et al. Phase I study of rigosertib, an inhibitor of the phosphatidylinositol 3-Kinase and polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer. Clin. Cancer Res. 2012, 18, 2048–2055.
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Hua, J.; Zhang, B.; Xu, J.; Yu, X. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/Snail signaling. Oncogene 2018, 37, 5843–5857.
- Sato, J.; Kimura, T.; Saito, T.; Anazawa, T.; Kenjo, A.; Sato, Y.; Tsuchiya, T.; Gotoh, M. Gene expression analysis for predicting gemcitabine resistance in human cholangiocarcinoma. J. Hepato-Biliary-Pancreat. Sci. 2011, 18, 700–711.
- Yin, T.; Wang, C.; Liu, T.; Zhao, G.; Zha, Y.; Yang, M. Expression of snail in pancreatic cancer promotes metastasis and chemoresistance. J. Surg. Res. 2007, 141, 196–203.
- Liu, W.-S.; Yan, H.-J.; Qin, R.-Y.; Tian, R.; Wang, M.; Jiang, J.-X.; Shen, M.; Shi, C.-J. siRNA directed against survivin enhances pancreatic cancer cell gemcitabine chemosensitivity. Am. J. Dig. Dis. 2008, 54, 89–96.
- Guo, Q.; Chen, Y.; Zhang, B.; Kang, M.; Xie, Q.; Wu, Y. Potentiation of the effect of gemcitabine by emodin in pancreatic cancer is associated with survivin inhibition. Biochem. Pharmacol. 2009, 77, 1674–1683.
- Guillermet-Guibert, J.; Davenne, L.; Pchejetski, D.; Saint-Laurent, N.; Brizuela, L.; Frugier, C.; Delisle, M.-B.; Cuvillier, O.; Susini, C.; Bousquet, C. Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol. Cancer Ther. 2009, 8, 809–820.
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115.
- Yabuuchi, S.; Pai, S.G.; Campbell, N.R.; de Wilde, R.F.; De Oliveira, E.; Korangath, P.; Streppel, M.M.; Rasheed, Z.A.; Hidalgo, M.; Maitra, A.; et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013, 335, 41–51.
- Espinoza, I.; Miele, L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013, 341, 41–45.
- Majidinia, M.; Alizadeh, E.; Yousefi, B.; Akbarzadeh, M.; Zarghami, N. Downregulation of notch signaling pathway as an effective chemosensitizer for cancer treatment. Drug Res. 2016, 66, 571–579.
- Du, X.; Zhao, Y.-P.; Zhang, T.-P.; Zhou, L.; Chen, G.; Wang, T.-X.; You, L.; Shu, H. Alteration of the intrinsic apoptosis pathway is involved in notch-induced chemoresistance to gemcitabine in pancreatic cancer. Arch. Med Res. 2013, 45, 15–20.
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461.
- Liang, Y.; Yang, L.; Xie, J. The role of the hedgehog pathway in chemoresistance of gastrointestinal cancers. Cells 2021, 10, 2030.
- Capurso, G.; Sette, C. Drug resistance in pancreatic cancer: New player caught in act. EBioMedicine 2019, 40, 39–40.
- Patzak, M.S.; Kari, V.; Patil, S.; Hamdan, F.; Goetze, R.G.; Brunner, M.; Gaedcke, J.; Kitz, J.; Jodrell, D.I.; Richards, F.M.; et al. Cytosolic 5′-nucleotidase 1A is overexpressed in pancreatic cancer and mediates gemcitabine resistance by reducing intracellular gemcitabine metabolites. EBioMedicine 2019, 40, 394–405.
- Donadelli, M.; Costanzo, C.; Beghelli, S.; Scupoli, M.; Dandrea, M.; Bonora, A.; Piacentini, P.; Budillon, A.; Caraglia, M.; Scarpa, A.; et al. Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochim. Biophys. Acta-Mol. Cell Res. 2007, 1773, 1095–1106.
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.; Kong, D.; Sarkar, F.H. Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2010, 8, 27–33.
- Xu, Z.; Vonlaufen, A.; Phillips, P.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.; Goldstein, D.; Pirola, R.C.; Wilson, J.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596.
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715.
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Castillo, C.F.-D.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856.
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Maitra, A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196.
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623.
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.-J.; Hotz, H.G. Epithelial to mesenchymal transition: Expression of the regulators snail, slug, and twist in pancreatic cancer. Clin. Cancer Res. 2007, 13, 4769–4776.
- Li, Y.; Kong, D.; Ahmad, A.; Bao, B.; Sarkar, F.H. Pancreatic cancer stem cells: Emerging target for designing novel therapy. Cancer Lett. 2013, 338, 94–100.
- Lee, C.J.; Dosch, J.; Simeone, D.M. Pancreatic cancer stem cells. J. Clin. Oncol. 2008, 26, 2806–2812.
- Abel, E.V.; Simeone, D.M. Biology and clinical applications of pancreatic cancer stem cells. Gastroenterology 2013, 144, 1241–1248.
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323.
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS ONE 2011, 6, e20636.
- A Rasheed, Z.; Matsui, W. Biologicaland clinical relevance of stem cells in pancreatic adenocarcinoma. J. Gastroenterol. Hepatol. 2012, 27, 15–18.
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769.
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell. Mol. Med. 2013, 17, 30–54.
- Delle Cave, D.; Di Guida, M.; Costa, V.; Sevillano, M.; Ferrante, L.; Heeschen, C.; Corona, M.; Cucciardi, A.; Lonardo, E. TGF-β1 secreted by pancreatic stellate cells promotes stemness and tumourigenicity in pancreatic cancer cells through L1CAM downregulation. Oncogene 2020, 39, 4271–4285.
- Zhou, Q.; Xia, S.; Guo, F.; Hu, F.; Wang, Z.; Ni, Y.; Wei, T.; Xiang, H.; Shang, D. Transforming growth factor-β in pancreatic diseases: Mechanisms and therapeutic potential. Pharmacol. Res. 2019, 142, 58–69.
- Zhao, J.; Liang, Y.; Yin, Q.; Liu, S.; Wang, Q.; Tang, Y.; Cao, C. Clinical and prognostic significance of serum transforming growth factor-beta1 levels in patients with pancreatic ductal adenocarcinoma. Braz. J. Med Biol. Res. 2016, 49, e5485.
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446.
- Simeone, D.M. Pancreatic cancer stem cells: Implications for the treatment of pancreatic cancer. Clin. Cancer Res. 2008, 14, 5646–5648.
- Mortezaee, K. Enriched cancer stem cells, dense stroma, and cold immunity: Interrelated events in pancreatic cancer. J. Biochem. Mol. Toxicol. 2021, 35, e22708.
- Niess, H.; Camaj, P.; Renner, A.; Ischenko, I.; Zhao, Y.; Krebs, S.; Mysliwietz, J.; Jäckel, C.; Nelson, P.J.; Blum, H.; et al. Side population cells of pancreatic cancer show characteristics of cancer stem cells responsible for resistance and metastasis. Target. Oncol. 2014, 10, 215–227.
- Xie, D.; Xie, K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015, 2, 133–143.
- Chen, X.; Zheng, P.; Xue, Z.; Li, J.; Wang, W.; Chen, X.; Xie, F.; Yu, Z.; Ouyang, X. CacyBP/SIP enhances multidrug resistance of pancreatic cancer cells by regulation of P-gp and Bcl-2. Apoptosis 2013, 18, 861–869.
- Chen, X.; Mo, P.; Li, X.; Zheng, P.; Zhao, L.; Xue, Z.; Ren, G.; Han, G.; Wang, X.; Fan, D. CacyBP/SIP protein promotes proliferation and G1/S transition of human pancreatic cancer cells. Mol. Carcinog. 2011, 50, 804–810.
- Takahashi, R.; Ijichi, H.; Sano, M.; Miyabayashi, K.; Mohri, D.; Kim, J.; Kimura, G.; Nakatsuka, T.; Fujiwara, H.; Yamamoto, K.; et al. Soluble VCAM-1 promotes gemcitabine resistance via macrophage infiltration and predicts therapeutic response in pancreatic cancer. Sci. Rep. 2020, 10, 1–13.
- Tadros, S.B.; Shukla, S.K.; King, R.J.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Lyudmyla, B.; Lazenby, A.J.; Berim, L.; et al. De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 2017, 77, 5503–5517.
- Qian, D.; Lu, Z.; Xu, Q.; Wu, P.; Tian, L.; Zhao, L.; Cai, B.; Yin, J.; Wu, Y.; Staveley-O’Carroll, K.F.; et al. Galectin-1-driven upregulation of SDF-1 in pancreatic stellate cells promotes pancreatic cancer metastasis. Cancer Lett. 2017, 397, 43–51.
- Masamune, A.; Satoh, M.; Hirabayashi, J.; Kasai, K.; Satoh, K.; Shimosegawa, T. Galectin-1 induces chemokine production and proliferation in pancreatic stellate cells. Am. J. Physiol. Liver Physiol. 2006, 290, G729–G736.
- Tang, D.; Yuan, Z.; Xue, X.; Lu, Z.; Zhang, Y.; Wang, H.; Chen, M.; An, Y.; Wei, J.; Zhu, Y.; et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int. J. Cancer 2011, 130, 2337–2348.
- Tang, D.; Wu, Q.; Zhang, J.; Zhang, H.; Yuan, Z.; Xu, J.; Chong, Y.; Huang, Y.; Xiong, Q.; Wang, S.; et al. Galectin-1 expression in activated pancreatic satellite cells promotes fibrosis in chronic pancreatitis/pancreatic cancer via the TGF-β1/Smad pathway. Oncol. Rep. 2018, 39, 1347–1355.
- You, Y.; Tan, J.-X.; Dai, H.-S.; Chen, H.-W.; Xu, X.-J.; Yang, A.-G.; Zhang, Y.-J.; Bai, L.-H.; Bie, P. MiRNA-22 inhibits oncogene galectin-1 in hepatocellular carcinoma. Oncotarget 2016, 7, 57099–57116.
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2020, 124, 333–344.
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838.
- Yang, X.; Yin, H.; Zhang, Y.; Li, X.; Tong, H.; Zeng, Y.; Wang, Q.; He, W. Hypoxia-induced autophagy promotes gemcitabine resistance in human bladder cancer cells through hypoxia-inducible factor 1α activation. Int. J. Oncol. 2018, 53, 215–224.
- Yin, H.; Yang, X.; Gu, W.; Liu, Y.; Li, X.; Huang, X.; Zhu, X.; Tao, Y.; Gou, X.; He, W. HMGB1-mediated autophagy attenuates gemcitabine-induced apoptosis in bladder cancer cells involving JNK and ERK activation. Oncotarget 2017, 8, 71642–71656.
- Boone, B.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.-C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; A Liotta, L.; et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410.
- Yang, M.-C.; Wang, H.-C.; Hou, Y.-C.; Tung, H.-L.; Chiu, T.-J.; Shan, Y.-S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 1–17.
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin. Cancer Res. 2020, 26, 3126–3134.
- Zhang, X.; Zhao, P.; Wang, C.; Xin, B. SNHG14 enhances gemcitabine resistance by sponging miR-101 to stimulate cell autophagy in pancreatic cancer. Biochem. Biophys. Res. Commun. 2019, 510, 508–514.
- AlMasri, S.S.; Zenati, M.S.; Desilva, A.; Nassour, I.; Boone, B.A.; Singhi, A.D.; Bartlett, D.L.; Liotta, L.A.; Espina, V.; Loughran, P.; et al. Encouraging long-term survival following autophagy inhibition using neoadjuvant hydroxychloroquine and gemcitabine for high-risk patients with resectable pancreatic carcinoma. Cancer Med. 2021, 10, 7233–7241.
- del Castillo, E.; Meier, R.; Chung, M.; Koestler, D.C.; Chen, T.; Paster, B.J.; Charpentier, K.P.; Kelsey, K.T.; Izard, J.; Michaud, D.S. The microbiomes of pancreatic and duodenum tissue overlap and are highly subject specific but differ between pancreatic cancer and noncancer subjects. Cancer Epidemiol. Biomark. Prev. 2018, 28, 370–383.
- Gleeson, F.C.; Jeraldo, P.; Levy, M.J.; Murphy, S.J.; Mendes-Soares, H.; Karagouga, G.; Mccune, A.F.; Deparedes, A.G.G.; Kipp, B.R.; Song, S.D.; et al. Composition, diversity and potential utility of intervention-naïve pancreatic cancer intratumoral microbiome signature profiling via endoscopic ultrasound. Gut 2021, 71, 441–443.
- Chung, M.; Zhao, N.; Meier, R.; Koestler, D.C.; Wu, G.; del Castillo, E.; Paster, B.J.; Charpentier, K.; Izard, J.; Kelsey, K.T.; et al. Comparisons of oral, intestinal, and pancreatic bacterial microbiomes in patients with pancreatic cancer and other gastrointestinal diseases. J. Oral Microbiol. 2021, 13, 1887680.
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980.
- McAllister, F.; Khan, A.W.; Helmink, B.; Wargo, J.A. The tumor microbiome in pancreatic cancer: Bacteria and beyond. Cancer Cell 2019, 36, 577–579.
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160.
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017, 170, 548–563.
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267.
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306.
- Wang, L.; Bi, R.; Yin, H.; Liu, H.; Li, L. ENO1 silencing impaires hypoxia-induced gemcitabine chemoresistance associated with redox modulation in pancreatic cancer cells. Am. J. Transl. Res. 2019, 11, 4470–4480.
- Tan, Z.; Xu, J.; Zhang, B.; Shi, S.; Yu, X.; Liang, C. Hypoxia: A barricade to conquer the pancreatic cancer. Cell. Mol. Life Sci. 2020, 77, 3077–3083.
- Abdalla, M.Y.; Ahmad, I.M.; Rachagani, S.; Banerjee, K.; Thompson, C.M.; Maurer, H.C.; Olive, K.P.; Bailey, K.L.; Britigan, B.E.; Kumar, S. Enhancing responsiveness of pancreatic cancer cells to gemcitabine treatment under hypoxia by heme oxygenase-1 inhibition. Transl. Res. 2019, 207, 56–69.
- Ma, J.; Weng, L.; Jia, Y.; Liu, B.; Wu, S.; Xue, L.; Yin, X.; Mao, A.; Wang, Z.; Shang, M. PTBP3 promotes malignancy and hypoxia-induced chemoresistance in pancreatic cancer cells by ATG12 up-regulation. J. Cell. Mol. Med. 2020, 24, 2917–2930.
- Yamasaki, A.; Yanai, K.; Onishi, H. Hypoxia and pancreatic ductal adenocarcinoma. Cancer Lett. 2020, 484, 9–15.
- Kasuya, K.; Tsuchida, A.; Nagakawa, Y.; Suzuki, M.; Abe, Y.; Itoi, T.; Serizawa, H.; Nagao, T.; Shimazu, M.; Aoki, T. Hypoxia-inducible factor-1α expression and gemcitabine chemotherapy for pancreatic cancer. Oncol. Rep. 2011, 26, 1399–1406.
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 1–15.
- Zhao, T.; Jin, F.; Xiao, D.; Wang, H.; Huang, C.; Wang, X.; Gao, S.; Liu, J.; Yang, S.; Hao, J. IL-37/ STAT3/ HIF-1α negative feedback signaling drives gemcitabine resistance in pancreatic cancer. Theranostics 2020, 10, 4088–4100.
- Zhang, X.; Kumstel, S.; Jiang, K.; Meng, S.; Gong, P.; Vollmar, B.; Zechner, D. LW6 enhances chemosensitivity to gemcitabine and inhibits autophagic flux in pancreatic cancer. J. Adv. Res. 2019, 20, 9–21.
- Ullah, A.; Leong, S.W.; Wang, J.; Wu, Q.; Ghauri, M.A.; Sarwar, A.; Su, Q.; Zhang, Y. Cephalomannine inhibits hypoxia-induced cellular function via the suppression of APEX1/HIF-1α interaction in lung cancer. Cell Death Dis. 2021, 12, 1–14.
- Kim, B.S.; Lee, K.; Jung, H.J.; Bhattarai, D.; Kwon, H.J. HIF-1α suppressing small molecule, LW6, inhibits cancer cell growth by binding to calcineurin b homologous protein 1. Biochem. Biophys. Res. Commun. 2015, 458, 14–20.
- Sato, M.; Hirose, K.; Kashiwakura, I.; Aoki, M.; Kawaguchi, H.; Hatayama, Y.; Akimoto, H.; Narita, Y.; Takai, Y. LW6, a hypoxia-inducible factor 1 inhibitor, selectively induces apoptosis in hypoxic cells through depolarization of mitochondria in A549 human lung cancer cells. Mol. Med. Rep. 2015, 12, 3462–3468.
- Zhang, Z.; Han, H.; Rong, Y.; Zhu, K.; Zhu, Z.; Tang, Z.; Xiong, C.; Tao, J. Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J. Exp. Clin. Cancer Res. 2018, 37, 291.
- Luo, G.; Xia, X.; Wang, X.; Zhang, K.; Cao, J.; Jiang, T.; Zhao, Q.; Qiu, Z. MiR-301a plays a pivotal role in hypoxia-induced gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2018, 369, 120–128.
- Han, Q.L.; Zhou, Y.H.; Lyu, Y.; Yan, H.; Dai, G.H. Effect of ribonucleotide reductase M1 expression on overall survival in patients with pancreatic cancer receiving gemcitabine chemotherapy: A literature-based meta-analysis. J. Clin. Pharm. Ther. 2017, 43, 163–169.
- Xie, H.; Jiang, W.; Jiang, J.; Wang, Y.; Kim, R.; Liu, X.; Liu, X. Predictive and prognostic roles of ribonucleotide reductase M1 in resectable pancreatic adenocarcinoma. Cancer 2012, 119, 173–181.
- Zhang, M.; Zhao, Y.; Zhang, Y.; Wang, D.; Gu, S.; Feng, W.; Peng, W.; Gong, A.; Xu, M. LncRNA UCA1 promotes migration and invasion in pancreatic cancer cells via the Hippo pathway. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 1770–1782.
- Zeng, R.; Dong, J. The hippo signaling pathway in drug resistance in cancer. Cancers 2021, 13, 318.
More