Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Gabriella Guelfi and Version 2 by Rita Xu.
P53 responds to stress in a variety of ways ranging from activating survival-promotion pathways to triggering programmed cell death to eliminate damaged cells. In physiological stress generated by any external or internal condition that challenges cell homeostasis, P53 exerts its function as a transcription factor for target genes or by regulating the expression and maturation of a class of small non-coding RNA molecules (miRNAs).
- P53
- miRNA
- physiological stress
1. P53 Is a Key Regulator of Cell Survival Pathways
Physiological cellular stress can be described as the outcome of the set of changes, related to the environment, that significantly disrupts cellular homeostasis by causing reversible damage to macromolecules such as proteins, DNA, RNA, and lipids. Cells in response to stress activate mechanisms such as growth arrest, repair or elimination of damaged macromolecules, changes in gene expression programs, not only to restore cellular homeostasis but also to enable adaptive homeostasis. Adaptive homeostasis enables biological systems to continually make short-term adjustments for optimal performance. If cellular damage is excessive, as caused by high doses of a stressor, programmed cell death (apoptosis) is triggered [1].
The tumor suppressor P53 protein, encoded by the TP53 gene, and its signaling pathway composed of hundreds of genes play a central role in maintaining homeostatic functions and adaptation to environmental fluctuations. The TP53 gene encodes a tumor-suppressive protein, activated by DNA damage, and also by other types of cellular stress. The functionally active P53 limits tumor development by limiting cell propagation into adverse conditions such as DNA damage, oncogene expression, and cellular stress. Even if growing normally, mice lacking P53 develop cancer before six months of age; however, there is growing evidence that P53 also contributes to pathologies other than cancer [2].
P53 is highly inducible by many stress signals, such as genotoxic stress, starvation, hypoxia, and oncogene activation. In non-stressed cells, the level of P53 is low, but during sustained stress signals, P53 drives irreversible programs of apoptosis or senescence to cull irreparably damaged or malignant cells for the ultimate benefit of the organism. In the event of low-level stress, P53 induces pro-survival homeostatic responses to protect genome integrity and to maintain viability in cells that suffer limited, reparable damage. The capability of P53 to coordinate multiple cell signaling pathways and determine cell fate has been the focus of numerous studies for many years [2].
As a transcription factor, the P53 protein mainly exerts its functions through transcriptional regulation by activating or repressing several functional clusters of genes. In response to a wide variety of intracellular and extracellular stress signals, P53 is mainly activated through post-translational modifications, leading to an increase in P53 half-life and its accumulation inside the cells. Thus, the transcription factor P53 binds to P53 response elements (P53RE) present in the early intron promoters of P53 target genes and induces the transcription of protein-coding or non-coding genes (microRNAs, etc.) involved in P53-mediated responses (Figure 1).
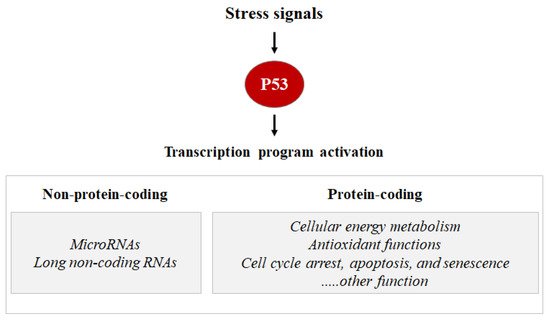
Figure 1. Stress signals can lead to P53 activation. Consequentially, the P53 can induce the transcription activation of coding proteins, ranging from the induction of cell cycle metabolism control, antioxidant functions apoptosis, senescence, cell cycle arrest, and DNA repair, or the P53 can induce the transcription activation of non-coding proteins such as microRNA and long non-coding RNAs.
In addition to transcription activation, P53 can also repress gene expression. The P53-dependent repression can occur directly through the binding of P53 to specific P53RE, or by recruiting co-repressors such as SIN3A. The P53–SIN3A complex targets HDAC1 (Histone Deacetylase 1) to P53-repressor promoters to create a chromatin environment that is not conducive to transcription. In addition, P53 can lock the binding sites for transcriptional activators that promote gene expression or the repression can occur through P53-dependent activation of non-coding RNAs that downregulate gene expression [3].
P53 protein plays a critical role in maintaining cellular energy metabolism by controlling oxidative phosphorylation and downregulation of glycolysis. Loss of P53 results in the deficiency of mitochondrial oxidative phosphorylation and enhanced glycolysis in both cultured cells and mouse models [4]. Another important function of P53 is to improve antioxidant defense by reducing ROS levels. To exert its antioxidant function, P53 induces a group of antioxidant genes, including a stress-inducible protein that regulates metabolism such as SESTRINs (SENS1, 2), TIGAR, GPX1, ALDH4, GLS2, and MnSOD, especially under conditions of non-stress or constitutive/mild stress, to lower ROS levels and prevent DNA damage [4]. Remarkably, in addition to antioxidant roles, P53 may exert a prooxidant effect via the upregulation of prooxidant genes, including PIG3, PIG6, FDXR, BAX, and PUMA [4]. Exposure of cells to high levels of ROS leads to oxidative stresses, which induce the P53 response (Figure 2).
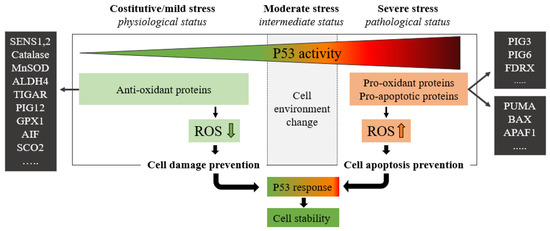
Figure 2. The green zone represents a low-risk state of constitutive/mild stress in which there is a homeostatic recovery. The dashed line represents the critical level of P53 activity; in this area, P53 attempts to ensure genome stability and cellular homeostasis by modulating P53-regulated genes. As the expression of P53 increases, the polychromatic triangle (top) changes from green to red, and cell damage risk increases due to the P53 expression level increment. During constitutive/mild stress, moderate stress, and severe stress, the cellular environment changes, and anti-oxidant or pro-oxidant proteins vary the ROS expressions with respect to the stress level. In mild stress, anti-oxidant proteins lower the level of ROS preventing cell damage. Conversely, in severe stress states, a high level of pro-oxidant and pro-apoptotic proteins increases ROS levels by inducing cell damage and apoptosis. ROS levels depend on P53 expression which in turn is stimulated by high ROS levels.
In response to prolonged or severe stress signals that induce irreparable damage, P53 drives irreversible apoptosis or senescence programs. P53 triggers apoptosis by the transcriptional induction of components of both the extrinsic and intrinsic death pathways, including BAX, FAS, NOXA, and PUMA [3]. In other cases, P53 responds to severe stress not with apoptosis but by inducing cellular senescence. Senescence is likely to result from the transcriptional activation of target genes such as CDKN1A, plasminogen activator inhibitor 1 (PAI1), and PML [3].
2. P53 Post-Transcriptional and Post-Translational Regulation
Several factors are able to specifically bind P53 mRNA or P53 protein to regulate P53 transcription or translation (Figure 3).
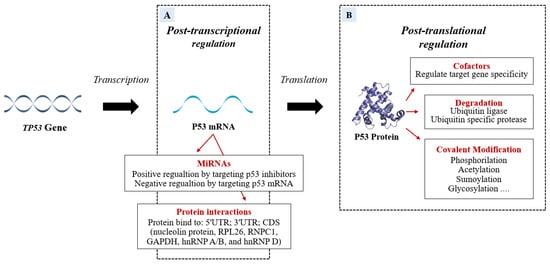
Figure 3. The P53 mRNA can be regulated post-transcriptionally (A) by miRNAs and RNA-binding proteins (RBPs) which modify the P53 transcriptional program in a stimulus-specific manner. The P53 miRNA regulation can be positive if miRNAs bind P53 inhibitors or can be negative if the miRNAs target is P53 mRNA. In addition to miRNA action, the P53 post-transcriptional program is also co-regulated, both negatively and positively, by RBPs that bind 5′UTR; 3′UTR; CDS of TP53. Some proteins modify P53 post-translationally. The figure (B) shows the P53-modifications of specific amino acid residues divided into Covalent Modification, Ubiquitination, and cofactors.
Important post-transcriptional regulators bind to P53 mRNA and regulate the protein levels to protect undamaged cells from the devastating consequences of abundant P53 and at the same time to ensure that damaged cells do not escape surveillance [5]. Recently, miRNAs have emerged as post-transcriptional regulators of P53. MiRNAs can be involved in physiological or pathological processes by regulating the abundance and/or activity of P53 (the role of miRNAs will be discussed in detail below). In response to cellular stresses, the interplay of P53 and post-translational modifications fine-tunes P53 activity to further prevent cellular harm. The post-translational level of the protein is primarily regulated by modifications such as ubiquitination, phosphorylation, acetylation, methylation, glycosylation, neddylation, and sumoylation.
The P53-ubiquitin ligase forms an autoregulatory loop resulting in lower P53 activity. Murine Double Minute 2 protein (MDM2), an E3 ubiquitin ligase, is the most important negative regulator of P53. Not only MDM2 but also other E3-ligases (such as COP1, PIRH2, and ARF-BP1) contribute to the control of P53 levels. MDM2 keeps at a low-level P53 in non-stress conditions through polyubiquitylation and proteasomal degradation. MDM2 binds to the transactivation domain of P53 and ubiquitylates its six C-terminal lysines, targeting it for proteasomal degradation. Together, P53 and MDM2 function in a negative feedback loop, P53, by binding to the MDM2 promoter, modulates the expression of MDM2 which in turn promotes the degradation of P53 and quenches cellular P53 activity [6].
In homeostatic conditions, P53 induces the transcription of the MDM2 gene maintaining low levels of P53 protein. However, upon cellular stress events, P53 protein levels rise due to a disruption of P53-mediated MDM2 transcription [7][8][7,8]. This negative feedback loop helps maintain the low cellular level of P53 under normal conditions and limits the duration and potency of the P53 activation under stress conditions [9]. Induction of P53 involves uncoupling it from its negative regulators, principally MDM2 and the related protein MDM4 (also known as MDMX). MDM4 has been identified as a protein sharing structural homology with MDM2, especially in the N-terminal P53 binding domain and both proteins have a role in maintaining low levels of P53 in normal cells. MDM4 suppresses P53 transcriptional activity by binding to its transactivation domain [10]. In addition, MDM4 downregulates P53 by enhancing MDM2-mediated degradation (Figure 4).
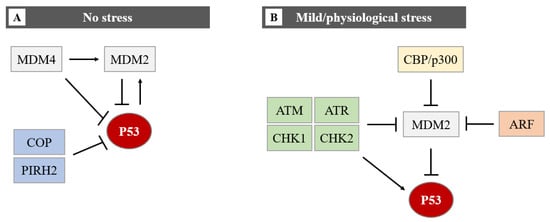
Figure 4. The regulation of the P53 protein level in cells in no stress and mild/physiological stress. During no stress (A), E3-ubiquitin ligases (MDM2, COP, and PIRH2) inhibit P53 activity through polyubiquitination and proteasome degradation. The function of P53 and MDM2 is regulated by a negative feedback loop. MDM4 can bind directly to P53 by repressing P53 transcriptional activity, or it can promote MDM2-mediated degradation of P53. The MDM2-P53 association promotes the degradation of P53 because physiological degradation of P53 is ubiquitin-dependent and MDM2 acts as a P53-specific E3 ubiquitin ligase. In conclusion, as MDM2 expression is positively regulated by P53, MDM2 drives P53 proteasomal degradation, it becomes apparent that MDM2 and P53 form an autoregulatory loop. In this loop, MDM2 restrains P53 levels under normal physiological conditions. Under mild/physiological stress states (B), MDM2 levels are kept low by ATM, ATR, CHK1 and CHK2, ARF, and CBP/p300. The low level of MDM2 induces the expression of P53. In the presence of mild/physiological stress, the level of these proteins increases.
The link between circular RNA (circRNAs) and P53 represents a further regulatory element of P53 whose implications may deserve great interest. CircRNAs are single-stranded non-coding RNAs, strongly involved in the regulation of P53 although there is information on their role in physiological stress. The circRNAs deriving from exons are generated from precursor mRNA mainly via a back-splicing process, in which the 3 ‘end of the exon ligates to the 5’ ends of its own forming a closed structure. The circRNAs can exert their function by binding and interacting with P53, to regulate the transcription process, acting with a mechanism similar to that of the ubiquitin ligase E3, MDM2, and COP1, resulting in the inhibition of P53 [11].
3. P53 Transcriptional Activation, and miRNA-mRNA Regulatory Network
The classical model for P53 activation consists of three sequential activating steps: stress-induced activation mediated by phosphorylation sequence-specific DNA binding, and transcriptional activation of the P53 target gene (Figure 5).
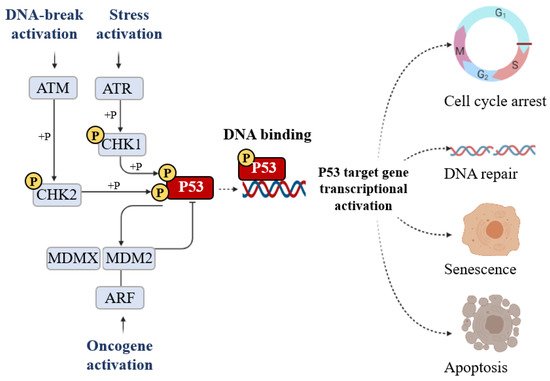
Figure 5. The signal that induces P53 activation is its phosphorylation (+P) which is triggered by DNA breakage activation (ATM), stress activation (ATR), and oncogene activation (ARF). Phosphorylation stabilizes P53 and promotes DNA binding. DNA-bound P53 then recruits the transcriptional machinery to activate the transcription of P53 target genes able of inducing cell cycle arrest, DNA repair, senescence, and apoptosis.
Post-translational modification is the major mechanism of P53 regulation and phosphorylation events strongly affect the transactivation function of P53. Following stress, P53 is phosphorylated at multiple residues, thereby modifying its biochemical functions, including P53 protein–protein interactions and DNA–protein interactions. Several residues at both the amino and carboxyl terminuses of P53 are phosphorylated or dephosphorylated in response to stress. The stabilization of P53 in cells experiencing stress is crucial for their homeostasis. The phosphorylation is a post-translational modification to P53 that affects its overall appearance and activity. P53 phosphorylation serves as a first focal point in the regulation of P53 activity and the presence of this modification is considered to be a marker of a stress response [12]. The convergence of multiple signals stimulating several P53 phosphorylations is required for a full transcriptional activation response. Following stress, P53 is phosphorylated at multiple residues, thereby modifying its biochemical functions required for increased activity as a transcription factor. To date, nine P53 phosphorylation sites (serine 20, 33, 46, 366, and 392 and threonine 81, 304, 377, and 387) have been detected in specific serine/threonine residues in the N- and C-terminal domains, all of which are able to induce apoptosis. The first crucial step of P53 stabilization is N-terminal phosphorylation at Ser15, Thr18, and Ser20, the latter being linked to P53-MDM2 regulation. Ser15 phosphorylation also triggers a sequential series of additional phosphorylation events in P53 (including phosphorylation of Ser9 -20, -46, and Thr18) that contribute further to P53 induction and activation. Ser15 phosphorylation is therefore an important focal point in the activation of P53 and in the association with the ubiquitin E3 ligases (p300 and MDM2) which mediate the ubiquitylation and proteasomal degradation of P53. In particular, a growing number of studies have indicated that phosphorylation at Ser46 was shown to play a fundamental role in the regulation of P53 apoptotic functions after DNA damage [13][14][13,14]. The biological role of the Ser20 site of P53 was identified as the stress-activated Ser20 kinases in the TAD1 transactivation domain [15]. An additional highly conserved phosphorylation site within the P53 C-terminus is the CK2 site on Ser392 whose phosphorylation by casein kinase 2 (CK2) stimulates the sequence-specific DNA-binding function of P53 [15].
P53 can be modified by phosphorylation by a broad range of kinases and the phosphorylation by cell cycle-dependent protein kinases suggests the possibility that the activity of P53 is regulated differentially during the cell cycle. Nevertheless, some studies stated that P53 can also be activated regardless of its phosphorylation state [16].
After activation of P53 in response to a stress signal, the likelihood of P53 binding to P53RE increases. The P53RE motifs are often located at 5′ of the gene or in the first or second intron of the gene regulated by P53. The P53REs have been characterized as the target gene regulatory regions [17]. In light of P53′s ability to bind DNA, the question of whether stress is required to induce binding between P53 and DNA is very interesting. Recent results demonstrate that P53 can bind the DNA of unstressed cells, but the inactivity of P53 is likely due to the repression of MDM2, and MDMX [18].
The homeostasis control of cells exposed to various stresses requires rapid and efficient reprogramming of mRNA translation, to preserve energy for repair and removal of stress damage [19]. Such shifts in stress-responsive protein synthesis the translation of specific mRNAs [20]. At the post-transcriptional level, via mRNA degradation and/or translational repression, more than 60% of total RNAs appear to be regulated by small single-stranded non-coding RNAs indicated as miRNAs (or microRNAs).
Research on miRNAs is one of the most widely discussed topics in science and medicine in the last decade. Bioinformatics tools and high-throughput sequencing contributed to the identification of numerous miRNAs and their potential gene targets. Over the last few decades, miRNAs have become very promising both in the area of biomarkers involved in the onset and progression of diverse biological anomalies and in the area of therapeutic approaches. There has been a drastic surge of interest in miRNA-based therapies developed to either suppress or restore the expression of disease-associated miRNAs. In circumstances where reduced miRNA expression drives the disease, miRNA mimics can be used to restore their expression and function [21]. MiRNAs are highly conserved across many vertebrate genomes [22]. MiRNAs offer the possibility of fast and economical post-transcriptional regulation of gene expression as they simultaneously target multiple transcripts without the need to synthesize proteins [23]. One miRNA regulates multiple mRNA targets, and one mRNA can be regulated by multiple miRNAs, highlighting the complexity of gene regulation by the miRNA network. MiRNAs negatively regulate gene expression by targeting the 3′UTRs of mRNAs. In addition to the cellular environment, there is a large proportion of miRNAs that migrate outside of it and can be found in biofluids including blood (plasma and serum), tears, saliva, urine, colostrum, breast milk, peritoneal fluid, amniotic fluid, cerebrospinal fluid, seminal fluid, synovial fluid, pleural fluid, bronchial lavage, and follicular fluid [24]. A plethora of recent studies has demonstrated that cell-free miRNA, called circulating miRNAs (cmiRNAs), represent the next generation of clinical, non-invasive, biomarkers for many pathologies [25]. In contrast to intracellular miRNAs, cmiRNAs are highly stable due to the kind of packaging that is essential to preventing the digestion of the miRNA by the RNases found in body fluids. Approximately 10% of cmiRNAs are secreted within microparticles such as exosomes, microvesicles, and apoptotic bodies, while the remaining 90% form complexes with RNA ligands such as lipoprotein complexes like high-density lipoprotein (HDL), Argonaute2 (Ago2) and nucleophosmin I (NPM 1) [21][26][21,26].
MiRNAs biogenesis is a complex process, which includes several crucial steps including transcription of primary (pri)-miRNAs; cleavage of pri-miRNAs to form precursor (pre)-miRNAs; and finally, cleavage of pre-miRNAs to form mature miRNAs [27] (Figure 6).
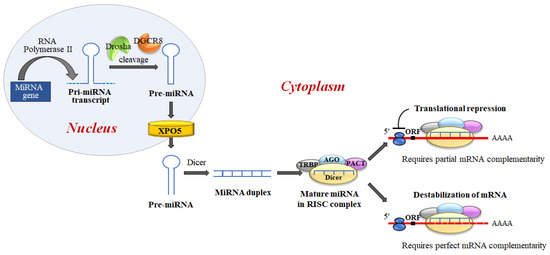
Figure 6. MiRNA biogenesis and miRNA regulation of gene expression. miRNAs are generated, in the nucleus, as primary transcripts (pri-miRNA) by RNA polymerase II/III. The miRNA precursor (pre-miRNA) is processed by Drosha, an RNase III enzyme, and by two molecules of a protein known as Pasha (DGCR8). DGCR8 contains an RNA-binding domain that binds the primary pri-miRNA to stabilize it for processing by Drosha. Drosha and DGCR8 homeostatically control miRNA biogenesis by an auto-feedback loop. After processing, pre-miRNAs, of approximately ~60–80 nt, are exported into the cytoplasm by Exportin-5 (XPO5). Next, the mature miRNA released by the Dicer endonuclease is assembled into the RNA-induced silencing complex (RISC). RISC directs its bound miRNA to untranslated regions of the partially complementary 3′mRNA, resulting in translation inhibition or targeted destruction of the mRNA. MiRNAs regulate target mRNA expression through two major mechanisms: translation blocking in the initiation step or the elongation phase; and removal of the polyA tail promoting deadenylase activity followed by mRNA degradation.
Disturbance of cellular homeostasis can interrupt each of the steps of miRNA biogenesis, leading to deregulation of the elaborate miRNA-controlled gene expression pathways, with the result that the cell is more susceptible to stress and its damaging outcomes. Emerging evidence suggests that stress directly influences not only miRNA quantity but also miRNA biogenesis [28][29][30][31][28,29,30,31].
In this complex network, miRNAs interact at multiple levels and the critical role that miRNAs play in the regulation of stress responses has only recently gained significant attention [29]. Interestingly, P53 regulates the expression of numerous miRNAs and in turn, it is a target of a set of miRNAs that represses P53 protein level and function, by forming a feedback loop with P53. MiRNAs can negatively regulate P53 protein levels through direct repression of P53 expression, or positively regulate P53 activity through the repression of several negative regulators of P53 [32].
Stress-induced variations in miRNA expression levels may partly explain the “stress hardening” phenomenon, clarified by Kultz et al. (2005) [33], in which miRNAs ensure the buffer of transcriptional peaks while maintaining a constant physiological level of gene expression [34]. This concept can be translated to stress resilience, in which the preconditioning of one stress increases tolerance to another stress [29]. As circulating miRNAs have a long half-life, their constant presence from the first exposure to a stressor allows the miRNA-mediated gene expression regulation to cope with future stressors exposure. However, stress is not always recoverable physiologically. If the stress becomes severe, it can induce changes in the miRNA sequence, modifications of miRNA end, “strand switching” [35] or the non-translating mRNAs and their RNA-binding proteins, aggregate into ribonucleoprotein granules to protect cellular mRNAs.