Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Maja Tomicic and Version 4 by Beatrix Zheng.
Glioblastoma multiforme (GBM) is a brain tumor characterized by high heterogeneity, diffuse infiltration, aggressiveness, and formation of recurrences. Patients with this kind of tumor suffer from cognitive, emotional, and behavioral problems, beyond exhibiting dismal survival rates. Current treatment comprises surgery, radiotherapy, and chemotherapy with the methylating agent, temozolomide (TMZ). GBMs harbor intrinsic mutations involving major pathways that elicit the cells to evade cell death, adapt to the genotoxic stress, and regrow.
- radiotherapy
- temozolomide
- acquired resistance
- mutational background
- apoptosis
- senescence
- stemness
- autophagy
1. Introduction
Primary tumors of the central nervous system (CNS) are, in respect to 5-year survival, among the top three most lethal cancers, only behind mesothelioma and pancreatic cancer [1][2][3][1,2,3]. However, since brain/spinal cord tumors comprise different entities, patients having the most aggressive form, glioblastoma multiforme (GBM, CNS WHO grade 4), survive an average 12–15 months from diagnosis [4], with a 5-year survival rate of 7.2%, surpassing the lethality of any other type of cancer [3]. Although gliomas have a low annual incidence (6 in 100,000 cases), currently it is increasing 3% per year [5]. Particularly if unresectable, many patients diagnosed with this type of cancer suffer from impairment in neurocognition, leading to behavioral, emotional, and other cognitive troubles, which often deteriorates with time. Long-term survivals continue to have significant symptom burden and care needs [6][7][6,7]. In 2021, the new WHO classification of CNS tumors was published [8][9][8,9]. In comparison to the WHO classification of 2016, this one relies on additional molecular markers introducing more accuracy and refinement in the definition of different classes of CNS tumors, including glioblastomas (GBM, CNS WHO grade 4). The 2021 classification no longer differentiates between primary and secondary GBMs. Particularly in respect to GBMs, it has a high clinical impact, affecting histologically lower astrocytoma grades that according to a specific molecular signature can be re-classified to GBMs. However, since there are not yet statistics in respect to correlation between molecular profiles, acquired therapy resistance, and corresponding clinical outcomes, as based on this new classification, information is rare, thwe researchers hhave predominantly concentrated on the molecular profiles, pathways, and clinical outcomes based on the 2016 WHO classification [10], but take into consideration the data with primary GBMs that correspond to the new classified ‘GBM, IDH-wildtype’ (CNS WHO grade 4) tumor entity.2. The Landscape of Genetic and Genomic Alterations in GBMs
The last decades of research have resulted in the identification of various chromosomes and genes that are frequently altered in GBMs. Considering the 2016 WHO classification, molecular genetic analyses have shown a different pattern of alterations in primary (90% of the GBM cases), secondary (10% of the cases), and treated relapsed tumors, [10]. The first well-established alterations were relative to isocitrate dehydrogenase IDH1/2 mutations, which became a factor for categorization and prognostication of these tumors [11][30]. The enzymes encoded by this group of genes work on chromosome remodeling/DNA methylation and metabolic signaling. Historically seen, mutated IDH1/2 allowed for clear differentiation between primary and secondary GBMs: it was frequent in secondary tumors and quite rare in primary or infant ones [12][13][31,32]. However, as already mentioned, the new 2021 WHO CNS classification does not differentiate between primary and secondary GBMs, and there is no entity such as ‘GBM, IDH-mutant’ [9][14][9,33]. Strictly speaking, the new classification defines GBM as ‘glioblastoma, IDH-wildtype’ (CNS WHO grade 4), with either one of the following altered molecular diagnostic markers [15][34]: TERT promoter mutation (~80%), simultaneous gain of chromosome 7 and loss of heterozygosity of chromosome 10q (+7/—0) [16][35], amplification and rearrangement of receptor tyrosine kinases, most commonly affecting EGFR (~50%) [15][17][34,36]. Thus, the new classified GBM, CNS WHO grade 4 entity reflects the primary GBM (WHO grade IV, according to the 2016 WHO classification), and overexpresses wild-type IDH1/2 protein [18][37]. The IDH1 gene mutations are often substitutions within exon 4 in the codon 132 and correspond to a more favorable outcome for the patient [19][38]. Likewise, there are also other quite rare but favorable IDH1/2 mutations [20][39]. According to the 2021 classification, they describe a new classified ‘astrocytoma, IDH-mutant’ tumor entity [8][9][8,9].It is important to underline that the new 2021 classification is a further evolvement of molecular profiling, combined with histologic tools, in diagnosis of the CNS tumors that was for the first time introduced by the breakthrough 2016 WHO classification, which integrated molecular and histologic features to refine classification of those tumors. Thus, other alterations in primary GBMs were recognized comprising the tumor suppressor genes (TSGs) TP53, RB1, CDKN2A (p16INKA4/p14ARF), and PTEN and the proto-oncogenes (POGs) EGFR, PDGFR, MET, CDK4, CDK6, CCND1, CCDN3, MDM2, MDM4, and MYCC [21][40] (Figure 12).
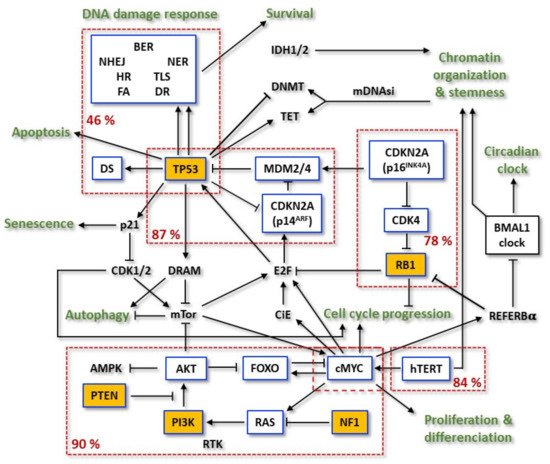
Figure 12. Flowchart of the most frequent mutations in the relevant pathways involved in primary GBM resistance. Within the boxes outlined in blue are significantly mutated genes (SMG) and in the yellow boxes are the driver-mutated genes. Dashed red boxes group mutated genes of the same pathway and display the relative mutation frequency of the pathway. Adapted from TCGA.
Different from the most TSGs, TP53 is rarely deleted in GBM [22][41]. The majority of its mutations are missense mutations and occur in the DNA binding domain (DBD), which leads to the inhibition of its transcriptional activity. The corresponding mutated protein is constitutively overexpressed and takes over oncogenic functions, not present in the wild-type (wt) form. These p53 mutants (mt) are called gain-of-function (GOF) p53mt [22][23][24][41,42,43]. On the other hand, mutations at other sites can bring out radically different cellular effects.
Further and more detailed discoveries were obtained with The Cancer Genome Atlas (TCGA) project, which aimed to catalogue cancer-causing genome alterations in large cohorts of human tumors using multi-dimensional analysis. GBM was the first cancer systematically studied by TCGA [21][40]. The first TCGA work analyzed 206 GBMs and, in addition to the previously described alterations, detected new significantly recurrent deletions involving NF1 and PARK2, and amplification of AKT3. PI3K complex activating missense mutations were also frequent. Rare, but informative events were also computed, such as FGFR2 and IRS2 amplifications and PTPRD deletion. Chromosome 7 gain (+7) (associated with EGFR amplification) and loss of heterozygosity of chromosome 10q (—10) (associated with PTEN loss) were among the most frequent genetic alterations [15][25][26][27][34,44,45,46]. For single nucleotide polymorphisms (SNPs) detection, 601 selected genes in 91 matched tumor-normal pairs (72 untreated and 19 treated cases) were sequenced. The results uncovered 453 validated non-silent somatic mutations in 223 unique genes. The background mutation was four times higher in treated tumors. Hypermutator phenotypes were described in 7 of the 19 samples treated with temozolomide (TMZ) and/or CCNU (lomustine) and were associated with mutations in at least one MMR gene; 48.5% of the tumors exhibited MGMT promoter methylation [17][36].
Later, TCGA expanded the analysis to more than 500 primary GBM samples [17][36]. Due to the nature of these tumors, IDH1 mutations were infrequent. Of those, 291 paired, tumor-normal genomic DNA samples were subjected to whole-exome sequencing (WES). Among the somatic mutations, 20,448 single nucleotide variants (SNVs), 39 dinucleotide mutations and 1153 small insertions and deletions (indels) were detected. Investigation of significantly mutated genes (SMGs) identified PTEN, TP53, EGFR, PIK3CA, PIK3R1, NF1, RB1, IDH1, PDGFRA, and the newly described LZTR1 as driver-mutated genes in GBMs (Figure 12). Interestingly, the protein encoded by LZTR1 is exclusively located at the Golgy network and, therefore, it is suspect to stabilize this complex. Other genes were also shown to have frequency above background, such as SPAT1, ATRX, GABRA6, and KEL. In the expanded analysis, the most common amplification events on chromosome 7 (EGFR/MET/CDK6), chromosome 12 (CDK4 and MDM2), and chromosome 4 (PDGFRA) were found to be in higher frequencies than previously reported. Single gene deletion targets included LRP1B, NPAS3, LSAMP, and SMYD3 [17][21][36,40].
Overall, 46% of the samples from a researchtudy were found to harbor at least one nonsynonymous mutation in a set of 161 genes functionally linked to chromatin organization. Telomerase reverse transcriptase (TERT) activating mutations and its subsequent higher mRNA expression were observed. Two hot spot mutations in the hTERT promoter region were mutually exclusive between each other and reached 84% of the cases [28][47]. On the other hand, hTERT mutations were also found to be mutually exclusive with ATRX mutations, concurrent with IDH1 and TP53. Receptor tyrosine kinases (RTKs) were found altered in 67.3% of the tumors: EGFR—57.4% (mutant variant vIII as the most common), PDGFRA—13.1%, MET—1.6% and FGFR2/3—3.2%. PI3K mutations were found in 25.1% of GBMs. Considering PI3K and PTEN, 89.6% had at least one alteration in the PI3K pathway, and 39% had two or more. TSG NF1, a GTPase-activating protein that negatively regulates the RAS/MAPK pathway, was deleted or mutated in 10% of cases and never co-occurred with BRAF mutations (2%). The p53 signaling pathway was found to be dysregulated in 85.3% of tumors, through mutation/deletion of TP53 (27.9%), amplification of MDM1/2/4 (15.1%), and/or deletion of CDKN2A (57.8%). The RB pathway was also disturbed by different events: 7.6% by direct RB1 mutation/deletion, 15.5% by amplification of CDK4/6, and the remainder via CDKN2A deletion [28][29][30][47,48,49].
Gene fusions and translocations in GBM have also been subject of investigation since they have the potential to create chimeric proteins with altered functions. Additionally, they can rearrange gene promoters that can lead to increased oncogene overexpression and activity or decreased expression of TSGs. These events can be very specific for the cancer type and can constitute a diagnostic marker. As an example, it is known that the translocation of the Philadelphia chromosome 9–22, which results in the BCR-ABL1 gene fusion, is characteristic of the chronic myeloid leukemia [31][50]. In GBM, it was found that FGFR3-TACC3 in-frame activating kinase fusion is a common marker and might play an important role in oncogenesis and cancer progression [32][51]. However, it is not quite specific to this kind of tumor.
Regarding DNA damage repair genes, it is known that these genes play key roles in maintaining genomic stability and the dysregulation of diverse repair functions is an important determinant of cancer risk, progression, and therapeutic response. Moreover, it was recently shown that DNA repair proteins may have a role beyond the level of DNA damage, revealing unexpected functions in transcriptional regulation, cancer stem cell (CSCs) signaling, and epithelial–mesenchymal transition (EMT) [33][52]. The prevalence of alterations in DNA repair genes was determined on 126 GBM samples. It was shown that numerous tumors harbored at least one alteration in core DNA repair genes. Five samples harbored alterations in the Fanconi Anemia (FA) pathway, three in the Non-Homologous End-Joining (NHEJ), three in Nucleotide Excision Repair (NER), one in BER, two in Translesion Synthesis (TLS), six in Homologous Recombination (HR), five in MMR, five in Damage Sensoring (DS), and 28 in DR [34][53]. In the assessment of the potential genomic consequences of these alterations, the authors also showed the association of TMZ signature (corresponding to COSMIC signature 11) with MGMT alterations [34][53]. The most frequent MGMT alteration found was the promoter methylation (92.4% of all alterations), leading to gene silencing. EXO5 silencing (via promoter methylation) was also frequent in GBMs (46%). When broken down to subtypes, EXO5 silencing was exclusively observed in IDH wild-type GBM. This observation suggests that EXO5 (5′ and 3′ bidirectional single-stranded DNA-specific exonuclease) may play a role in the pathogenesis of subsets of these cancers. For instance, recently it was shown that functional deficiency of this DNA repair gene plays a role in prostate tumorigenesis [34][35][53,54].
Another group of genes that has been implicated in cancer initiation, progression, and resistance to therapy is the group of the autophagy-related genes (ARG). Autophagy is an important homeostasis mechanism, which degrades and recycles components of the cells in lysosomes, providing materials and energy [36][55]. The autophagic pathway shares numerous components with the RTK/PI3K/AKT signaling. However, beyond these common players, autophagy-related proteins functionally control and regulate the autophagosome formation, including initiation, nucleation, elongation of the membrane, maturation of autophagosomes, trafficking, docking, fusion with lysosome membranes, and degradation of intra-autophagosomal contents in a hierarchical manner [37][56]. In this context, a significant correlation of some autophagy (ATG) gene polymorphisms and glioma susceptibility was reported [38][57]. Moreover, when analyzing expression data of 986 patients, a signature containing 14 ARGs able to predict GBM patients’ prognosis was obtained by researchers [39][40][58,59]. Of those, the expression of MTMR4, LENG9, P4HB, PCIRG1, HSPA5, DRAM1, CTSD, S100A8, and CCL2 increased with signature risk score. Additionally, patients with this signature, classified as high-risk, had more malignant characteristics than those classified as low risk [39][58].
Cancer progression also encompasses gradual loss of a differentiated phenotype and acquisition of progenitor-like, stem-cell-like features. Undifferentiated primary tumors are more likely to metastasize and to be resistant to current therapies [41][60]. Malta and co-workers defined two signatures to quantify stemness based on a multi-platform analysis: one was reflective of epigenetic features (mDNAsi, DNA methylation-based stemness index) and the other of gene expression (mRNAsi, mRNA expression-based stemness index) [42][61]. For GBMs, they found a strong association between high pathologic grade and recently published glioma subtypes: mDNAsi was highest in highly aggressive GBMs. In addition, high mDNAsi was strongly associated with mutations in NF1 and EGFR and infrequent mutations in IDH1, TP53, CIC, and ATRX. The authors also found a negative correlation between higher stemness indices and reduced leukocyte fraction and lower PD-L1 expression. These findings reveal that GBMs might be less susceptible to immune checkpoint blockade treatments due to insufficient immune cell infiltration or preexisting downregulation of the PD-L1 pathway, which makes further inhibition ineffective [42][61]. Indeed, large-scale phase III clinical trials with immune checkpoint inhibitors did not increase GBM patients’ survival [43][44][62,63].
Using bioinformatic tools to find common putatively actionable alterations in a set of 33 types of cancer (including GBMs), it was found that across the entire data set, CDKN2A deletions (13%), PIK3CA mutations (12%), MYC amplifications (8%), and K-RAS mutations (7%) were the most common [45][46][64,65]. Of those, CDKN2A loss may predict sensitivity to CDK4/6 inhibitors. The BRAF V600R sequence variant, which confers sensitivity to vemurafenib in melanomas, was only detected in 1.7% of the GBMs. MYCN mRNA expression was also found upregulated in GBMs. In an investigation on the potential differences with respect to MYC-associated pathways, GBMs were found to harbor the MYC canonical signature with respect to DNA replication/repair and chromatin. It could also be shown that they exhibit the MYCN signature associated with genes related to neuronal function and development [45][46][47][64,65,66]. Because the studies from the TCGA consortium have the largest sample sizes and the most comprehensive, up to date systematic analyses (from which many other studies have been out seeded), it is reasonable to assume the frequencies of those alterations as putative statistics for GBMs.
All these molecular features have led researchers to propose a taxonomic approach to GBM, dividing it into four molecular subtypes: classical, neural, pro-neural, and mesenchymal. However, the methodological analyses of the samples used for this characterization comprised not only the bona fide tumor cells but also the tumor-associated nonmalignant cells. A more stringent analysis, considering the separation of malignant (GBM) and nonmalignant (glial) cells led to a state-of-the-art classification into three subtypes: classical, pro-neural, and mesenchymal, suggesting the former neural subtype would arise from contamination of the samples with non-malignant cells [48][49][50][51][67,68,69,70]. Moreover, as already mentioned, recent re-classification of tumors from the CNS by the WHO 2021 [8][9][52][8,9,71] has taken more molecular features as diagnostic criteria into consideration. In this regard, the most relevant change refers to low-grade astrocytomas and IDH-mt astrocytomas. Molecular profiling showed that the overwhelming majority of histologically lower CNS WHO grade 2 and grade 3 diffuse astrocytomas exhibiting the IDH-wt status, shared signature in genomic alterations (either EGFR amplification, or concurrent +7/—10, or TERT promoter mutation) and clinical outcomes with the former primary GBM, WHO grade IV or currently classified GBM, CNS WHO grade 4, suggesting those tumors were only under-represented GBMs [53][72]. Additionally, IDH-mt diffuse astrocytomas are now considered entities per se (astrocytoma, IDH-mutant, WHO grade 2, 3, 4). These changes have recently been summarized [14][33].