Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Anton Goncharov and Version 2 by Rita Xu.
Adenosine-to-inosine RNA editing is a system of post-transcriptional modification widely distributed in metazoans which is catalyzed by ADAR enzymes and occurs mostly in double-stranded RNA (dsRNA) before splicing. This type of RNA editing changes the genetic code, as inosine generally pairs with cytosine in contrast to adenosine, and this expectably modulates RNA splicing.
- RNA editing
- RNA splicing
- ADAR
1. Introduction: Adenosine-to-Inosine RNA Editing by ADAR Enzymes
Before the transcriptional machinery of animal cells provides processing of newly synthesized messenger RNAs by the spliceosome, another important post-transcriptional RNA modification occurs. This is RNA editing by one or more enzymes of the Adenosine Deaminase, RNA-dependent (ADAR) family. Generally, ADARs bind double-stranded secondary structures on RNA and then hydrolytically deaminate adenine bases nearby, which results in the formation of hypoxanthine bases as part of newly formed inosine nucleosides instead of adenosines, respectively [1]. ADAR enzymes bind long, regularly paired RNA duplexes, with multiple adenosines modified as a result; they also bind short, imperfect RNA duplexes, and specific adenosine residues are edited nearby, providing specific editing [2]. Adenosine-to-inosine RNA editing has been described for most types of RNA, including messenger and non-coding RNA species [3].
What are the important changes that are introduced by adenosine conversion to inosine? The fundamental chemical result of this modification is that the exchange of an amino to an oxy moiety changes the ability to form hydrogen bonds [4]. Inosine residues are complementary to cytosine in RNA, not to uracil, mimicking guanosine [5]. Thus, the secondary structure of a transcript is modified after editing; in particular, double-stranded structures are destroyed. In coding parts of mRNA, inosines serving as guanosines in triplets in some cases recode translated protein sequences [6]. A fixed number of amino acid substitutions is generated by this type of mRNA modification, some of which are well characterized and have functional impacts that will be discussed below.
Since the discovery of RNA editing of dsRNA in the Xenopus frog embryo by Bass and Weintraub as far back as 1987 [7], there has been growing interest in the enzymes catalyzing this reaction and in the pathways that the editing is involved in. Soon after the original discovery, it was found that ADAR orthologs are a metazoan innovation and that they are found in many animals, from sponges to mammals [8]. Among the animal kingdom, a number of ADAR-encoding genes vary. Among the well-studied examples in the context of RNA editing, the fruit fly has a single gene for the enzyme, as cephalopods and mammals possess two and three ADAR genes, respectively [9].
Structurally, the enzymes contain one or more dsRNA-binding domains, a catalytic deaminase domain and, optionally, a Z-DNA-binding domain with a function unknown until recently (Figure 1) [8]. A major activity of ADARs in organisms in which they have been studied is to deaminate adenosine to inosine in dsRNA sites. This reaction serves two canonical functions of RNA editing. First is the disruption of dsRNA structures. In most known animals, the level of dsRNA in the cytoplasm is subject to innate immunity surveillance by specific sensors that react to excess dsRNA as a potential viral genomic load. Correspondingly, RNA editing has immunosuppressive action and, e.g., works as negative feedback during type I interferon response in mammals, where this induces a production of the cytoplasmic isoform of ADAR1, p150, from an alternative transcription start, which is longer than the constitutive p110 isoform [10]. A second functional impact of RNA editing by ADARs is protein recoding. As inosine pairs more preferably to cytosine in complementary RNA strains, the editing of residues in exonic mRNA parts may cause a fixed number of amino acid substitutions dictated by the genetic code [11], some of them being not conservative and structurally important, such as glutamine to arginine or tyrosine to cysteine. During ontogenesis, recoded proteins are shown to be produced with or even instead of their genomically encoded prototypes and, in a few cases, functional roles have been attributed to these substitutions [6][12][13][6,12,13]. Two above-mentioned functions—immunosuppressive and protein-recoding functions—are, in most cases, specifically performed by different ADAR paralogs, such as ADAR1 and ADAR2 in mammals and cephalopods, respectively, except in the insects, where ADAR1 was lost and functional diversity is reached by alternative splicing and self-editing of the ADAR2 ortholog [14]. In mammals and cephalopods, ADAR1 isoforms contain Z-DNA-binding domains, whereas ADAR2 isoforms and its insect ortholog lack these domains (Figure 1) [9].
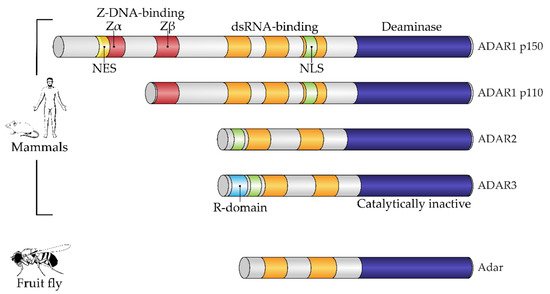
Figure 1. Domain architecture of mammalian and fruit fly ADAR family members. All proteins of the ADAR family share a common domain architecture and include a variable number of dsRNA-binding domains (dsRBDs) and a catalytic deaminase domain. In addition, human ADAR1 isoforms and their orthologs also contain Z-DNA-binding domains. A constitutive isoform, ADAR1 p110, contains the so-called Zβ domain, which adopts a fold similar to Zα domains but is not able to bind a Z-form of nucleic acid. Interferon-inducible ADAR1 p150 has an extended N-terminus containing a Zα domain, which harbors a nuclear export signal (NES). ADAR3, a catalytically inactive member of the family, additionally contains an R-domain. Fruit flies have a single Adar, which is most similar to mammalian ADAR2. NES, nuclear export signal; NLS, nuclear localization signal; R-domain, arginine-rich domain. Figure composed based on [15], with amendments.
In metazoans, significant variation is observed in the protein-recoding function of ADARs. Thus, the exonic sequences of many insects and cephalopods have been shown to be enriched by recoded sites [16][17][18][16,17,18], in contrast to mammals, in which the number of recoded sites is relatively low. It is thought that invertebrates use RNA editing to recode their proteins, mostly in neural and glandular tissues, providing better flexibility during ontogeny accompanied with metamorphosis [19] and for better adaptation to changing ambient temperatures [20][21][20,21]. Two complementary theories for the explanation of the evolutionary significance of protein recoding via ADAR editing have been suggested [2]. First, recoding diversifies proteoforms in addition to genomic variation and alternative splicing, which facilitates adaptation during ontogeny and changing circumstances, as mentioned above. Changes in nucleic acid codes that are introduced by RNA editing are even considered as RNA mutations. Indeed, in the context of the RNA world which presumably preceded DNA genomes in early living systems, these RNA mutations represented a driving force in early evolution [22]. A second theory, which does not contradict the first one, states that RNA editing restores functional sequences after deleterious genomic mutations [23]. Evolutionary aspects of A-to-I RNA editing have recently been reviewed in more detail [2][9][24][25][2,9,24,25].
In correspondence with transcriptome data, a proteome-wide analysis of recoding by ADAR activity has shown that in insects and cephalopods many more proteins have these substitutions than in mammals [16][26][27][16,26,27]. In humans, not more than tens of recoding cases may be identified even in deep proteomic data, the recoded sites being found mostly in the brain [28] and in various tumors [29].
As mentioned above, ADAR enzymes—ADAR1, in particular—are active in most human tissues. Is the RNA modification that they enable essential for survival? In humans, mutations in ADAR1 cause an orphan disease, one of the forms of Aicardi–Goutières syndrome, associated with enhanced interferon responses and inflammation in vital organs [30]. Severe autoimmune responses observed in this syndrome illustrate the vital immunosuppressive function of ADAR1 [31]. Notably, the deadly deficit of ADAR1 in mice may be rescued by knockout of the dsRNA sensor, MDA5 [32]. Similar to ADAR1, mutations of ADAR2 are harmful and, in most cases, fatal during early development [33], despite this isoform being expressed preferentially in the central neural system and in blood vessels in mammals. Very rare cases of patients with this orphan disease, caused by ADAR2 mutations and still unnamed, were recently described [34]. The deficit was accompanied by seizures combined with serious neurological disorder. An essential function of ADAR2 is associated with a single recoded site in the glutamate ionotropic receptor AMPA type subunit 2 (GRIA2), where glutamine is substituted by arginine in position 607 [35]. The resulting substitution decreases conductivity of the corresponding ion channel, which is vital for the developing brain. In ADAR2-deficient mice, a genomic knock-in of a corresponding arginine residue rescued the transgenic animals [36].
The role of A-to-I RNA editing in human cancer has been widely discussed and reviewed elsewhere [37]. Different cancers are characterized by differential expression of ADARs and, correspondingly, various levels of RNA editing [38]. Thus, brain cancers are characterized by low RNA editing [39], whereas hyper- or misediting is reported for some thyroid, head and neck, lung and breast cancers [40]. According to its known immunoregulatory role, the ADAR1 enzyme is generally considered as an oncogenic agent that aids tumors in resisting immunity [38][41][38,41]. On the contrary, mRNA editing of the ADAR2 isoform may form neo-antigens through recoding protein sequences and has been attributed a tumor-suppressive ability [42].
Protein recoding via ADAR2 editing of corresponding transcripts was shown to be proapoptotic in esophageal squamous cell carcinoma. Recoding of the IGF-binding protein IGFBP7 downregulated cancer cell proliferation, presumably by inhibition of the Akt pathway [43]. Moreover, the tumor-suppressive action of ADAR2 may be associated, inter alia, with its recently found role in DNA repair. In double-strand DNA breaks, DNA–RNA hybrids are formed which interfere with the DNA-end resection machinery repairing the breaks. ADAR2 has been shown to disrupt the hybrids catalytically interacting locally with BRCA1 and SETX genomic caretakers [44].
Evidence has accumulated that, besides their action via canonical transcript-editing functions, ADARs regulate cancer by editing-independent mechanisms. In glioblastoma, a proto-oncogenic methyltransferase, METTL3, enhanced production of ADAR1 protein via methylation (m6A) of its transcript. Experimental silencing of ADAR1 in this cancer attenuated proliferation, probably through down-regulation of the CDK2 transcript, independently of enzymatic function [45].
2. ADAR1 and Type I Interferon Signaling in Cancer
As mentioned above, an important major function of human ADAR1 is to inactivate intrinsically transcribed dsRNA, which, at excessive levels, can cause innate immune reactions. Transcription of short interspersed nuclear elements, such as Alu elements of the human genome, is a major source of dsRNA in transcripts, as they have potential for intramolecular coupling, with the long dsRNA bands formed resembling some viral genomes. For example, an unprocessed pre-mRNA contains, on average, ten to twenty Alu repeats in its intronic parts [46]. When dsRNAs are transferred to the cytosol, which can occur, for example, as a result of splicing dysregulation, innate immunity sensors, normally responsible for viral nucleic acid recognition, are activated [47]. A major sensor of dsRNA is Melanoma Differentiation-Associated protein 5 (MDA5) which belongs to the group of RIG-I-like receptors [48]. MDA5 recognizes and binds dsRNAs of several hundred nucleotides. After its binding, the activated receptor, via a well-described signaling pathway, induces type I interferon gene expression and secretion of these cytokines [49]. Type I interferons, in turn, act as paracrine and autocrine regulators and, through their receptors, activate the expression of a large pool of specific interferon-stimulated genes (ISGs) [50]. The type I interferon pathway plays a leading role in antiviral response. One group of ISG products arrests the translation of cell and viral proteins, among which protein kinase RNA-activated (PKR) is well described [51]. Eventually, continuous type I interferon signaling arrests cell division and even may lead to apoptosis, which is relevant to cancer cells. Thus, splicing dysregulation in cancer cells, as well as viral attack, such as oncolytic viral treatment, may be beneficial for disease control. At the same time, type I interferons stimulate expression of some genes encoding immunosuppressive proteins as part of a negative feedback loop in response to the main interferon effects [52]. As mentioned above, type I interferon signaling provokes the alternative splicing of ADAR1 transcripts. As a result, a long p150 isoform of this enzyme is produced which contains a cytosol transport signal and, in cytosol, can deactivate dsRNAs as part of the negative feedback response to interferons. Its deaminating enzymatic activity decreases levels of dsRNA in the cytoplasm and, accordingly, immunostimulatory signaling by dsRNA sensors (Figure 2). In addition to RNA editing, the p150 isoform contains a Z-DNA/RNA-binding domain (Zα) which is shown to bind and stabilize RNAs with the Z-RNA conformation in stress granules formed during type I interferon response [53]. The conservation of mRNA in stress granules makes it possible to continue protein translation after its temporary arrest caused by PKR activity [54].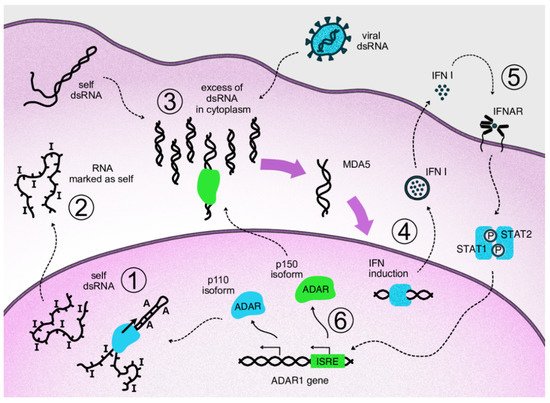
Figure 2. Negative feedback loop of cytosolic ADAR1 isoform acting on type I interferon signaling via inactivation of dsRNA. Repetitive elements of the genome produce transcripts capable of forming double-stranded RNA structures. In the nucleus, these structures undergo ADAR-mediated deamination (1). The cell perceives modified transcripts as self, so their appearance in the cytoplasm does not lead to activation of the dsRNA reactive pathway (2). If levels of dsRNA in cytoplasm are increased as a result of viral attack or overproduction of self dsRNA, e.g., via overexpression of Alu repeats, corresponding sensors are activated, such as MDA5 and RIG-1 (3). Activation of the sensors triggers antiviral innate immunity through transcription factors that initiate type I interferon expression (4). Autocrine and paracrine interferon signaling leads to the transcriptional activation of interferon-stimulated genes via phosphorylated STAT1/STAT2 complexes binding specific genomic ISRE elements (5). In the ADAR1 gene, interferon signaling switches expression to the p150 isoform, which migrates to the cytosol and edits excess dsRNA, thus acting on type I interferon signaling as part of a negative feedback loop (6).
Table 1. Small-molecule splicing and ADAR editing inhibitors.
| Family | Compound | Target/Effect | Clinical Trials |
|---|---|---|---|
| RNA splicing inhibitors | |||
| Spliceostatins | Spliceostatin A | Target SF3B1, block A complex assembly | - |
| Sudemycin D6 | - | ||
| Meayamycin B | - | ||
| Pladienolides | FD-895 | - | |
| Pladienolide B | - | ||
| E7107 | + | ||
| H3B-8800 | + | ||
| Herboxidiene | Herboxidiene | - | |
| GEX | - | ||
| 18-Deoxyherboxidiene | - | ||
| Isoginkgetin | Isoginkgetin | Prevents binding of U4/U5/U6 tri-snRNP to the A complex |
- |
| Histone deacetylase (HDAC) inhibitors | Suberoylanilide hydroxamic acid | B complex | - |
| Splitomicin | - | ||
| Dihydrocoumarin | - | ||
| Histone acetyltransferase inhibitors | Garcinol | A complex | - |
| AA | B complex | - | |
| BA3 | - | ||
| Kinase inhibitors | Diospyrin | H/E complex | - |
| Chlorhexidine | - | ||
| TG003 | - | ||
| Antibiotics | Chlortetracycline | Early spliceosome assembly | - |
| Streptomycin | - | ||
| Erythromycin | C complex | - | |
| Sulfanilamide | Indisulam | Degradation of splicing factor RBM39 | + |
| E7820 | - | ||
| CQS | - | ||
| Tasisulam | + | ||
| ADAR RNA-editing inhibitors | |||
| Adenosine analogs | 8-Azaadenosine | ADAR1 | - |
| 8-Chloroadenosine | - | ||
| Adenine analog | Erythro-9-(2-hydroxy-3-nonyl) adenine hydrochloride (EHNA) | ADA, a metabolic adenosine deaminase; EHNA also inhibited some ADAR2 editing events | - |