Gangliosides (glycosphingolipids containing one or more sialic acids) are highly expressed in neural tissues in vertebrates, and four species (GM1a, GD1a, GD1b, GT1b) are predominant in mammalian brains. GM3 is the precursor of each of these four species and is the major ganglioside in many nonneural tissues. GM3 synthase (GM3S), encoded by
ST3GAL5
gene in humans, is a sialyltransferase responsible for synthesis of GM3 from its precursor, lactosylceramide.
ST3GAL5
mutations cause an autosomal recessive form of severe infantile-onset neurological disease characterized by progressive microcephaly, intellectual disability, dyskinetic movements, blindness, deafness, intractable seizures, and pigment changes.
- gangliosides
- GM3 synthase
- glycosyltransferase
1. Introduction
Gangliosides—glycosphingolipids (GSLs) that contain one or more sialic acids—are fundamental components of cell membrane microdomains involved in dynamic regulation of various cell physiological processes [1]. Ganglioside biosynthesis is initiated by the addition of a glucose residue to ceramide to form glucosylceramide (GlcCer); this process is catalyzed by GlcCer synthase (GlcCerS, encoded by UGCG gene) (Figure 1). Similarly, a galactose residue can be added to ceramide to form galactosylceramide (GalCer), catalyzed by GalCer synthase (GalCerS, encoded by UGT8). Synthesis of GlcCer occurs on the cytosolic surface of Golgi, whereas GalCer synthesis occurs on the luminal surface of endoplasmic reticulum [2][3]. Sulfation of GalCer by cerebroside sulfotransferase (CST, encoded by GAL3ST1 gene) generates sulfatide SM4, a major lipid component of myelin sheath in both the central nervous system (CNS) and peripheral nervous system (PNS). GlcCer is galactosylated by lactosylceramide (LacCer) synthase (LacCerS, encoded by B4GALT5 or B4GALT6) to form LacCer, the precursor of a variety of ganglioside species and other types of GSLs. GM3 synthase (GM3S, encoded by ST3GAL5 gene) is a sialyltransferase that adds a sialic acid residue to LacCer to initiate synthesis of a- and b-series gangliosides. GD3 synthase (GD3S, encoded by ST8SIA1 gene) is another sialyltransferase that catalyzes formation of disialoganglioside GD3. GM2 synthase (GM2S, encoded by B4GALNT1 gene) adds N-acetylgalactosamine (GalNAc) to LacCer, GM3, or GD3 to generate (respectively) GA2, GM2, or GD2. Gangliosides are expressed in essentially all vertebrate tissues and cells, most abundantly in the nervous system. GM1, GD1a, GD1b, and GT1b in particular are the predominant ganglioside species in mammalian brain tissues [4].
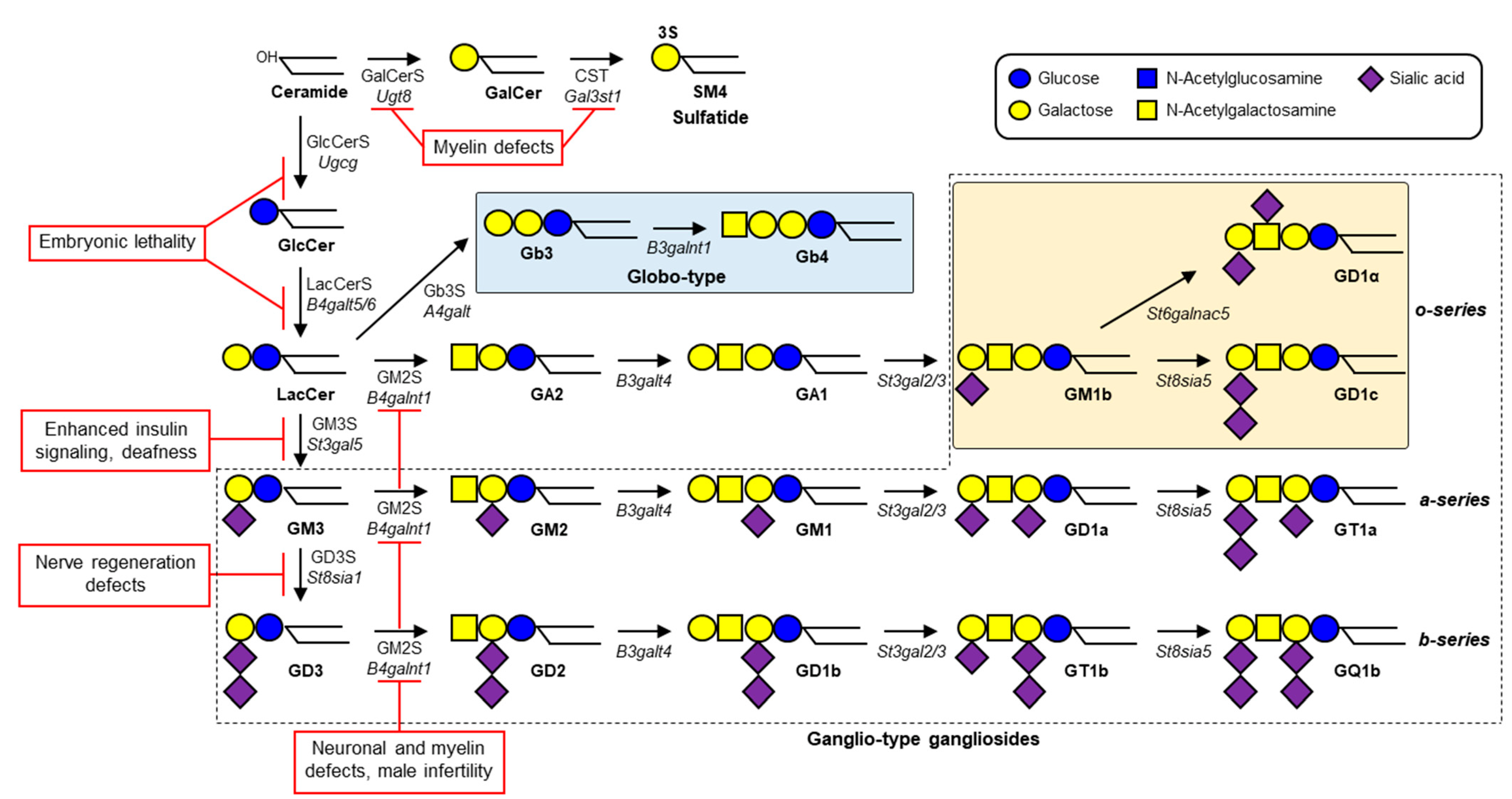
2. Human GM3S Deficiency
GM3S deficiency was first reported as an autosomal recessive infantile-onset epilepsy syndrome associated with developmental stagnation and blindness in Old Order Amish [5]. Biallelic pathogenic ST3GAL5 variants led to disrupted synthesis of a- and b-series gangliosides, and consequently to severe infantile-onset neurological disorders characterized by progressive microcephaly, intellectual disability, choreoathetosis, blindness, deafness, intractable seizures, and/or pigment changes [5][6][7][8][9][10][11][12][13]. These patients appeared normal at birth, but all had psychomotor developmental delays, and most displayed consistent features of movement disorders and/or epilepsy. A “salt and pepper” syndrome sometimes seen in African American patients results from altered dermal pigmentation (hyper- or hypopigmented skin maculae) at various locations [8]. The human GM3S protein consists of 418 amino acids and includes sequences for L-(large), S- (small), and VS- (very small) sialyl motifs, which are highly conserved in mammalian sialyltransferases [14]. A nonsense variant, R288X, detected in numerous Amish GM3S-deficiency patients, two French patients, and three Pakistani patients results in a truncated protein that lacks S and VS. motifs, and was therefore predicted to be nonfunctional [5][7][10]. Plasma GSL analysis of the Amish patients revealed a total absence of GM3 and its downstream derivatives, whereas increased levels were observed for LacCer (the direct substrate of GM3S), o-series gangliosides (e.g., GM1b), and globosides [11]. Several missense variants are located in L or S motifs: E355K (found in African American patients with salt and pepper syndrome), C195S and G201R (found as compound heterozygotes in Korean patients), and G342S (found in an Italian patient). In vitro GM3S assay for LacCer using homogenate of HEK293T cells transfected with mutated ST3GAL5 constructs revealed an absence of enzymatic activity in these variants [12]. A recent study revealed four more novel variants: missense variants G247D and H389R, nonsense variant R344X, and stop-loss variant X419RextX38, which disrupts the stop codon and causes 37-codon extension of the reading frame [13]. Plasma GSL analysis of patients with these four variants showed significant reduction of GM3 levels; however, GM3 was still present, as well as residual GM2—which was not observed in Amish patients with the R288X variant. The pathogenic mechanisms of these variants will be clarified by in vitro GM3S assay and analysis of intracellular localization and/or stability of ST3GAL5 protein. Two patients, homozygous for the above stop-loss variant, had the highest plasma GM3 levels among the studied patients, and the least severe phenotype [13]. Plasma GM3 levels thus appear to be inversely related to severity of GM3S-deficiency disease.
References
- Jin-Ichi Inokuchi; Kei-Ichiro Inamori; Kazuya Kabayama; Masakazu Nagafuku; Satoshi Uemura; Shinji Go; Akemi Suzuki; Isao Ohno; Hirotaka Kanoh; Fumi Shishido; et al. Biology of GM3 Ganglioside. Progress in Molecular Biology and Translational Science 2018, 156, 151-195, 10.1016/bs.pmbts.2017.10.004.
- Hein Sprong; Boudewijn Kruithof; Richtje Leijendekker; Jan Willem Slot; Gerrit van Meer; Peter van der Sluijs; UDP-Galactose:Ceramide Galactosyltransferase Is a Class I Integral Membrane Protein of the Endoplasmic Reticulum. Journal of Biological Chemistry 1998, 273, 25880-25888, 10.1074/jbc.273.40.25880.
- D Jeckel; A Karrenbauer; K N Burger; Gerrit van Meer; F Wieland; Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. Journal of Cell Biology 1992, 117, 259-267, 10.1083/jcb.117.2.259.
- Ronald L. Schnaar; The Biology of Gangliosides. Advances in Carbohydrate Chemistry and Biochemistry 2018, 76, 113-148, 10.1016/bs.accb.2018.09.002.
- Michael Simpson; Harold Cross; Christos Proukakis; David A Priestman; David C A Neville; Gabriele Reinkensmeier; Heng Wang; Max Wiznitzer; Kay Gurtz; Argyro Verganelaki; et al.Anna PrydeMichael A PattonRaymond A DwekTerry D ButtersFrances PlattAndrew H Crosby Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nature Genetics 2004, 36, 1225-1229, 10.1038/ng1460.
- Heng Wang; Alicia Bright; Baozhong Xin; J.R. Bockoven; Amy S. Paller; Cutaneous dyspigmentation in patients with ganglioside GM3 synthase deficiency. American Journal of Medical Genetics Part A 2013, 161, 875-879, 10.1002/ajmg.a.35826.
- Konstantina Fragaki; Samira Ait-El-Mkadem; Annabelle Chaussenot; Catherine Gire; Raymond Mengual; Laurent Bonesso; Marie Bénéteau; Jean Ehrland Ricci; Valérie Desquiret-Dumas; Vincent Procaccio; et al.Agnes RotigV Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency. European Journal of Human Genetics 2012, 21, 528-534, 10.1038/ejhg.2012.202.
- Luigi Boccuto; Kazuhiro Aoki; Heather Flanagan-Steet; Chin-Fu Chen; Xiang Fan; Frank Bartel; Marharyta Petukh; Ayla Pittman; Robert Saul; Alka Chaubey; et al.Emil AlexovMichael TiemeyerRichard SteetCharles E. Schwartz A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Human Molecular Genetics 2013, 23, 418-433, 10.1093/hmg/ddt434.
- Jin Sook Lee; Yongjin Yoo; Byung Chan Lim; Ki Joong Kim; Junghan Song; Murim Choi; Jong-Hee Chae; GM3 synthase deficiency due toST3GAL5variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype. American Journal of Medical Genetics Part A 2016, 170, 2200-2205, 10.1002/ajmg.a.37773.
- Eliza Gordon-Lipkin; Julie S. Cohen; Siddharth Srivastava; Bruno P. Soares; Eric Levey; Ali Fatemi; ST3GAL5-Related Disorders: A Deficiency in Ganglioside Metabolism and a Genetic Cause of Intellectual Disability and Choreoathetosis. Journal of Child Neurology 2018, 33, 825-831, 10.1177/0883073818791099.
- Lauren E. Bowser; Millie Young; Olivia K. Wenger; Zineb Ammous; Karlla W. Brigatti; Vincent J. Carson; Teresa Moser; James Deline; Kazuhiro Aoki; Thierry Morlet; et al.Ethan M. ScottErik PuffenbergerDonna L. RobinsonChristine HendricksonJonathan SalvinSteven GottliebAdam D. HeapsMichael TiemeyerKevin A. Strauss Recessive GM3 synthase deficiency: Natural history, biochemistry, and therapeutic frontier. Molecular Genetics and Metabolism 2019, 126, 475-488, 10.1016/j.ymgme.2019.01.013.
- Rossella Indellicato; Rossella Parini; Ruben Domenighini; Nadia Malagolini; Maria Iascone; Serena Gasperini; Nicoletta Masera; Fabio Dall’Olio; Marco Trinchera; Total loss of GM3 synthase activity by a normally processed enzyme in a novel variant and in all ST3GAL5 variants reported to cause a distinct congenital disorder of glycosylation. Glycobiology 2019, 29, 229-241, 10.1093/glycob/cwy112.
- Solveig Heide; Marie-Line Jacquemont; David Cheillan; Michel Renouil; Marilyn Tallot; Charles E. Schwartz; Juliette Miquel; Marc Bintner; Diana Rodriguez; Françoise Darcel; et al.Julien BurattiDamien HayeSandrine PassemardDomitille GrasLaurence PerrinYline CapriBénédicte GérardAmélie PitonBoris KerenChristel Thauvin-RobinetYannis DuffourdLaurence FaivreCharlotte PoeAnne PervilléDelphine HéronJulien ThévenonLionel ArnaudEric LeGuernLorita La SelvaAnnalisa VetroRenzo GuerriniCaroline NavaCyril Mignot GM3 synthase deficiency in non-Amish patients. Genetics in Medicine 2021, 24, 492-498, 10.1016/j.gim.2021.10.007.
- James C. Paulson; Christoph Rademacher; Glycan terminator. Nature Structural & Molecular Biology 2009, 16, 1121-1122, 10.1038/nsmb1109-1121.