The NLR family apoptosis inhibitory proteins (NAIP)-NLR family caspase-associated recruitment domain-containing protein 4 (NLRC4) inflammasome is important for mounting an immune response against Gram-negative bacteria. NLRC4 is activated through NAIPs sensing type 3 secretion system (T3SS) proteins from Gram-negative bacteria, such as Salmonella Typhimurium. Mutations in NAIPs and NLRC4 are linked to autoinflammatory disorders in humans.
- NOD-like receptors
- NLR
- NLRC4
- inflammasome
- innate immunity
- PANoptosis
- PANoptosome
- infection
- NAIP
- Gram-negative bacteria
- IL-1β
- IL-18
- Salmonella
- caspase
- gasdermin
1. Introduction
Innate immunity is the front-line defense mechanism to protect the host from various pathogenic and sterile insults. The innate immune system is triggered by pathogen-associated molecular patterns (PAMPs), which are components of infectious agents, and damage-associated molecular patterns (DAMPs), which are released during cellular or tissue damage. PAMPs and DAMPs are detected by germline-encoded host sensors called pattern recognition receptors (PRRs) [1][2][1,2], and this detection and subsequent response are critical for host survival. Based on their localization, PRRs are classified as membrane-bound PRRs and cytoplasmic PRRs. Membrane-bound PRRs include Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), whereas cytoplasmic PRRs include nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs), and RIG-I-like receptors (RLRs). NLRs recognize a diverse array of ligands which can be from self or non-self, including pathogens. After sensing the ligands, some of the NLR family members assemble an inflammasome, which is a cytosolic multiprotein complex. Inflammasome assembly drives the activation of caspase-1, which processes IL-1β and IL-18 to produce their active forms and cleaves gasdermin D (GSDMD) to trigger a form of inflammatory cell death known as pyroptosis [3][4][5][3,4,5]. Due to their importance in inflammation and host response to pathogens, mutations in NLRs are associated with human autoimmune and autoinflammatory disorders. Although NLR family pyrin domain (PYD)-containing 3 (NLRP3) is the best-characterized inflammasome sensor, several other sensors, including NLRP1, NLR apoptosis inhibitory protein (NAIP)-NLR family caspase recruitment domain (CARD)-containing protein 4 (NLRC4), NLRP6, NLRP9, absent in melanoma 2 (AIM2), and Pyrin, can also form inflammasomes and participate in regulating the host immune and inflammatory response [6][7][8][6,7,8].
NLRC4 plays a critical role in detecting Gram-negative bacteria in the cytoplasm, and it was initially discovered and called IPAF (ICE protease-activating factor) for its ability to activate caspase-1 [9]. NLRC4 features a three-domain structure: an amino-terminal CARD, a central nucleotide-binding domain (NACHT), and a carboxy-terminal leucine-rich repeat domain (LRR) (Figure 1).

NLRC4 can associate with pro–caspase-1 directly through CARD-CARD interactions, which triggers the processing and activation of caspase-1 [10]. Additionally, the adaptor molecule ASC (apoptosis-associated speck-like protein containing a CARD), encoded by the gene Pycard, can also facilitate this interaction. Activated NLRC4 can associate with ASC and colocalizes with the ASC-containing speck during Salmonella Typhimurium infection [11][12][11,12]. ASC contains a PYD and a CARD (Figure 1); its CARD is critical for optimal caspase-1 recruitment into the speck to allow its activation and proteolytic cleavage of pro–IL-1β and pro–IL-18 [11][12][13][11,12,13]. Further, activated caspase-1 drives proteolytic cleavage of the pore-forming protein GSDMD, allowing the N-terminus of GSDMD to oligomerize in the host cell membrane which results in pore formation that causes pyroptosis and the release of cytokines and alarmins (Figure 2). NAIPs act as upstream sensors for NLRC4 inflammasome assembly. They contain an N-terminal baculovirus IAP-repeat (BIR) domain, a central NACHT, and a carboxy-terminal LRR [6] (Figure 1). NAIPs are crucial for detecting bacterial ligands in the cytoplasm, and their association with NLRC4 triggers NAIP-NLRC4 inflammasome activation [6] (Figure 2).
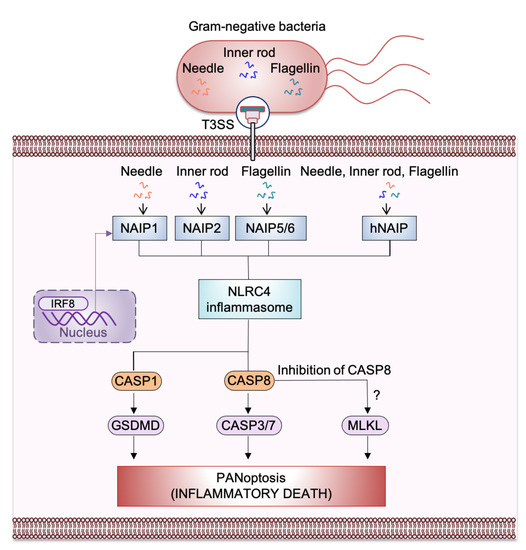
Figure 2. Mechanism of nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family apoptosis inhibitory protein (NAIP)-NLR family caspase recruitment domain-containing protein 4 (NLRC4) inflammasome-mediated PANoptosis, inflammatory cell death. Ligands from Gram-negative bacteria such as needle and inner rod protein of the Type 3 secretion system (T3SS) can be detected by murine NAIP1 and NAIP2, respectively, whereas flagellin can be detected by murine NAIP5/NAIP6. The single human NAIP (hNAIP) detects all the T3SS proteins such as needle, inner rod protein, and flagellin. After detecting T3SS proteins, NAIP associates with NLRC4 to induce activation of the NAIP/NLRC4 inflammasome. This activation results in inflammasome assembly and the cleavage and activation of caspase-1 (CASP1). Activated CASP1 triggers gasdermin D (GSDMD)-mediated pyroptosis. NLRC4 also activates caspase-8 (CASP8), which goes on to activate caspase-3/-7 (CASP3/7) to trigger apoptosis. Activation of the necroptotic pathway has also been observed during S. Typhimurium infection; this may occur through inhibition of CASP8. NAIPs are transcriptionally induced by interferon regulatory factor 8 (IRF8).
2. Role of NLRC4 in Host Defense
The innate immune function of the NLRC4 inflammasome has been extensively studied during bacterial infections, especially the foodborne bacterium S. Typhimurium. Studies have documented that Nlrc4-deficient animals are more susceptible to S. Typhimurium infection and have increased bacterial loads in the cecum, liver, and spleen compared with control animals [11][14][15][16][17][11,28,29,30,31], suggesting that NLRC4 is indispensable to protect the host from S. Typhimurium infection. However, similar studies have demonstrated that Nlrc4 knockout mice have no difference in bacterial load compared to wildtype mice after S. Typhimurium infection [13][17][18][13,31,32]. Functional redundancies between NLRC4 and NLRP3 could be the reason for these conflicting results. Indeed, later studies have shown that Nlrc4 and Nlrp3 double knockout mice are highly susceptible to S. Typhimurium and have increased bacterial loads in the spleen, liver, and mesenteric lymph nodes compared with control animals [13]. Other potential factors that could be contributing to the conflicting results observed in studies of NLRC4 during S. Typhimurium infection include differences in the bacterial strain used for infection, the route of bacterial administration, the genetic background of the mice used, and the gut microbiota of mice housed in different animal facilities. The intricate relationship between the gut microbiota and inflammasome activation has made it clear that using littermate controls is essential for obtaining reliable results from in vivo experiments [19][20][33,34]. Littermates are preferable to antibiotic depletion of the native microbiome, as treating mice with antibiotics can cause gut injury which was found to be associated with sepsis-like disease and the systemic spread of a multidrug-resistant E. coli pathobiont which can activate the NAIP5-NLRC4 inflammasome [21][35], further confounding results. Despite these conflicting findings, it is clear that NLRC4 is important in sensing bacterial ligands during S. Typhimurium infection [11][14][15][16][17][11,28,29,30,31].
Similar to S. Typhimurium, another enteric bacterial pathogen Citrobacter rodentium causes increased pathology and hyper-susceptibility in mice deficient in Nlrp3, Nlrc4, and Casp1, suggesting a critical role for NLRC4 in host defense against C. rodentium [22][36]. In addition to the role of NLRC4 in immune cells from the hematopoietic compartment during this infection, NLRC4 activation in the non-hematopoietic cellular compartment also occurs, especially in gut epithelial cells. Transplanting wildtype bone marrow cells to Nlrc4−/− mice using a bone marrow chimera technique does not control the C. rodentium pathogen load, suggesting NLRC4 is important in the non-hematopoietic compartment [23][37]. Follow-up studies confirmed the essential functions of NLRC4 in the non-hematopoietic compartment by using tissue-specific gene knockout mice. Similar results have been observed with other pathogens. For example, mouse gut epithelial-specific deletion of Naip1–6 leads to an increased pathogen load after Salmonella infection [15][29]. Later studies showed that the expulsion of infected enterocytes relies on NLRC4 by using gut epithelium-specific Nlrc4 knockout mice [16][30].
Apart from its importance in the host response to these enteric pathogens, NLRC4 also plays a crucial role in eliminating non-enteric bacteria. The NAIP-NLRC4 inflammasome can be activated by the flagellated pneumonia-causing bacteria Legionella pneumophila. Nlrc4-deficient mice fail to clear L. pneumophila upon nasal infection, while wildtype mice can clear the pathogen [24][25][26][27][38,39,40,41]. Similar to the importance of NLRC4 during infection with L. pneumophila, Nlrc4-deficient mice infected with other Legionella species, including L. micdadei, L. bozemanii, L. gratiana, and L. rubrilucens, are more susceptible and have increased bacterial burden compared with wildtype infected mice [25][26][27][28][39,40,41,42]. Later studies have also demonstrated the importance of NLRC4 during P. aeruginosa infection. Nlrc4-deficient macrophages are markedly resistant to P. aeruginosa-induced cell death and have reduced secretion of IL-1β. P. aeruginosa isolates express the effector molecule exoenzyme U (ExoU), which is capable of inhibiting caspase-1–driven proinflammatory cytokine production [29][43]. Upon exposure to P. aeruginosa, Nlrc4-deficient mice have an increased bacterial burden in the bronchoalveolar lavage, but not in lung tissues when compared to control animals [30][44].
In contrast to these protective roles of NLRC4 during several Gram-negative bacterial infections, NLRC4 can also contribute to pathogenesis. Helicobacter pylori can cause chronic infection and lead to gastric ulcers and gastric adenocarcinomas. NLRC4 is crucial for IL-18 production from both human and murine gastric epithelial cells upon exposure to H. pylori. Nlrc4-deficient mice have reduced inflammation and control the bacterial burden more successfully than wildtype infected mice do [31][45], highlighting the pathogenic role NLRC4 can play during infection.
Overall, these studies show that the NAIP-NLRC4 inflammasome has dual roles during bacterial infection. NLRC4 protects the host against certain pathogens, such as Salmonella, Citrobacter or Legionella, but its activation may also be pathogenic by triggering strong inflammation during some infections, such as during Helicobacter infection.