Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Christine Varon and Version 2 by Camila Xu.
The Hippo signalling pathway is one of the most crucial and complex ones in physiology, and there is no doubt that the regulatory mechanisms it possesses are various. The role of this signalisation process in tissue homeostasis makes it keen to lead to cancerous processes when dysregulated.
- gastric cancer
- hippo
- YAP
- TAZ
- cancer stem cells
1. The Hippo Pathway
1.1. Overview
The Salvador-Warts-Hippo pathway (SAV1-LATS1/2-MST1/2 in mammals) is the key regulator of organ size and tissue homeostasis in physiology. Ever since its discovery in Drosophila [1][2][1,2], described as “hippopotamus phenotype” (contributing to its name), where its deregulation induces a dramatic tissue overgrowth, this evolutionary conserved pathway has been described in divers physiological and pathological processes; from cell growth, proliferation, cell-cell contact, cell density and cell polarity control, stemness, shear stress, tissue homeostasis, and repair and regeneration, to cancer [3][4][5][6][7][8][9][10][11][12][13][3,4,5,6,7,8,9,10,11,12,13]. Genetic screenings for genes involved in cell growth led to the discovery of its different members [5][6][14][5,6,14]. This highly conserved pathway in mammals is made up of two central groups of elements driving its role: 1) a serine-threonine kinase core comprising of Mammalian Sterile-20 such as 1/2 (MST1/2) and Large tumour suppressor 1/2 (LATS1/2) kinases and, 2) a transcriptional module containing Yes-associated protein (YAP) [15][16][17][15,16,17] and/or a Transcriptional co-activator, with a PDZ binding motif (WWTR1 or TAZ) (Figure 1).
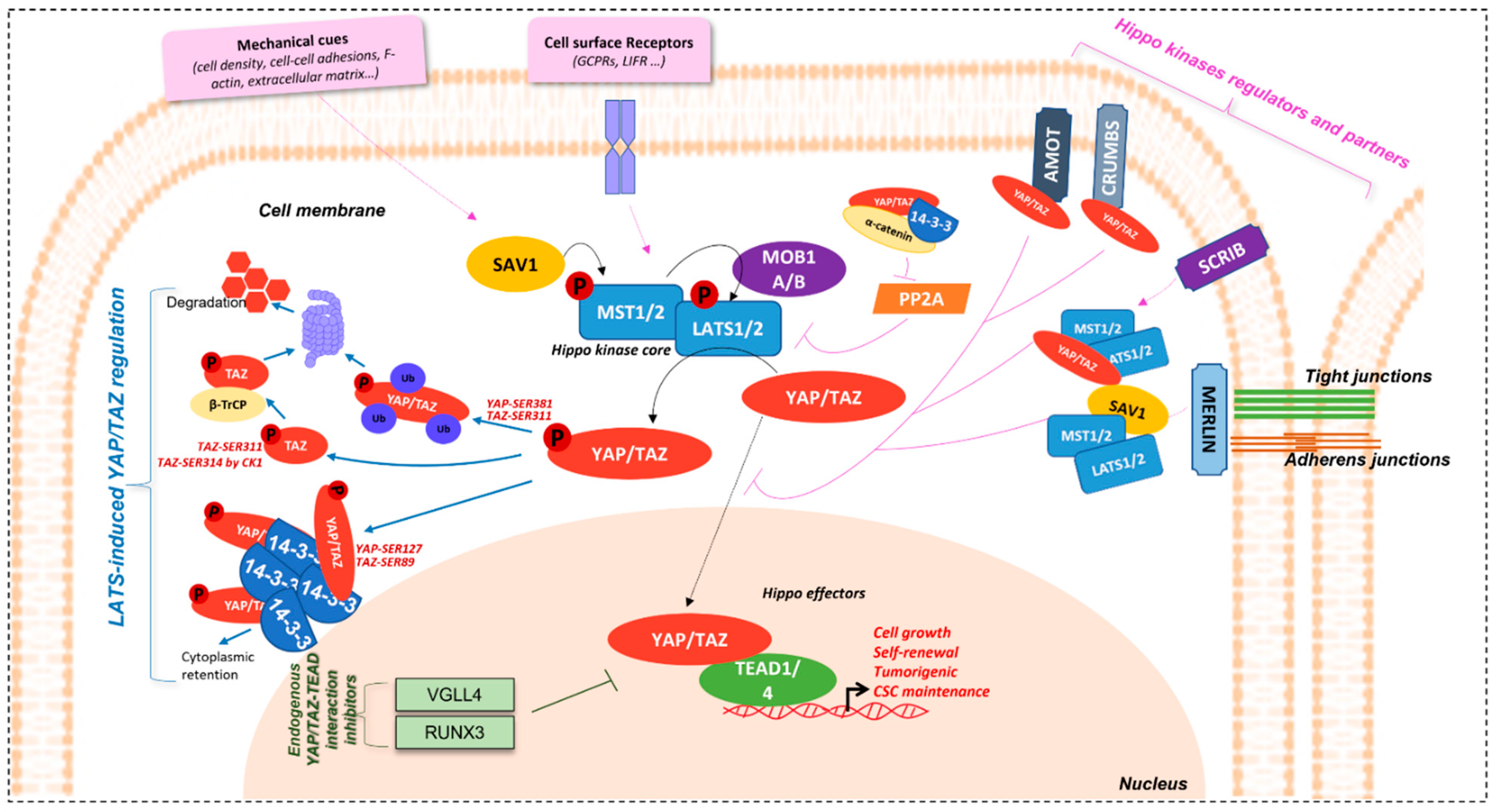
Figure 1. Hippo/YAP/TAZ-TEAD signalling pathway. Schematical representation of Hippo signalisation network. Regulation of YAP/TAZ effectors by LATS are represented by the blue arrows, endogenous YAP/TAZ-TEAD inhibitors in green, and Hippo kinases regulators and partners in pink.
YAP/TAZ are transcriptional co-activators. They do not possess DNA-binding motifs and need DNA-binding partners to accomplish their roles. The major ones are TEA domain family member 1–4 (TEAD1-4), also known as the Transcriptional enhancer factor (TEF) in mammals, and Scalloped (Sd) in Drosophila [18][19][18,19]. TEAD fixation by YAP/TAZ is significant in the mediation of its biological functions. Screening of the human transcription factor library, coupled with luciferase transcription reporter assays and/or yap co-transfection, allowed the identification of TEADs as the major actor of YAP binding to gene promoters and of YAP-induced transcriptional activity [6][19][6,19].
Studies show that YAP/TAZ are the effectors carrying the functional activity of the pathway. Mutations of Yorkie (Yki), YAP/TAZ ortholog in Drosophila, lead to decrease in proliferation, while the gigantism phenotype and liver tissue expansion are observed with its overexpression, coinciding with the phenotype observed upon Hippo kinases mutation. YAP deletion in mice decreases cell overgrowth phenotype caused by a lack of MST1/2 [5][8][5,8]. Active YAP mutant transgenic expression or dysregulation, through deletion of Hippo kinase members at the embryonic stage, leads to the hyperproliferation of cardiomyocytes and heart enlargement [20][21][22][23][20,21,22,23].
In addition, YAP1 overactivation in the intestines causes enlargement of the pool of multipotent undifferentiated progenitor cells, which undergo differentiation once YAP induction is ceased [8][11][8,11]. YAP inhibition also re-establishes cell-contact inhibition. Strict control of these effectors is, thus, required for proper tissue homeostasis.
1.2. Regulation Mechanisms
Hippo pathway regulators intervene at different levels for the maintenance of proper tissue homeostasis (Figure 1).
1.2.1. YAP/TAZ Effectors Regulation
Hippo effectors are, indeed, tightly controlled by upstream members of the pathway, the hippo kinases, and their regulatory partners, Salvador Family WW Domain Containing Protein 1 (SAV1) and MOB Kinase Activator 1A/B (MOB1A/B), playing a role in the activation of MST1/2 and LATS1/2, respectively. Phosphorylation cascade of members of this kinase core induces the phosphorylation of YAP/TAZ effectors and, thus, represses their nuclear localisation and activity [3][6][9][18][24][3,6,9,18,24]. This is demonstrated in hippo kinases inhibition models, where nuclear accumulation of the effectors is observed [9][25][9,25].
Among the inhibition mechanisms are cytoplasm sequestration or nuclear exclusion mechanisms. YAP/TAZ are active in the nucleus and their nuclear accumulation is, thus, elemental for their function as co-transcription factors. Hippo kinase LATS1/2 phosphorylate YAP and TAZ on Ser-127 (p-YAPSER127) and Ser-89 (p-TAZSER89) residues, respectively, allowing their binding to 14-3-3 proteins, thus retaining them in the cytoplasm [3][18][3,18]. In addition, 14-3-3 proteins knock-down (KD) induces nuclear accumulation, which is consistent with this observation.
Other 14-3-3 proteins-independent mechanisms also exist [25]. For example, LATS1/2 also phosphorylates YAP and TAZ on Ser-381 and Ser-311, respectively, modulating their protein stability, by inducing their polyubiquitination, and their addressing to the proteasome, for degradation. Indeed, studies have shown that TAZ Ser-311 phosphorylation by LATS1/2 can prime it to be further phosphorylated on Ser-314 on their phosphodegron motif by Casein kinase 1 (CK1), allowing interaction with β-TrCP, and, then, with SCF E3 ubiquitin ligases, to be directed to the proteasome for degradation [10][26][10,26]. This effector inhibition mechanism is absent in the Drosophila model.
Furthermore, YAP/TAZ contains a COOH-terminal domain, allowing interaction with proteins having PDZ binding domains [3][18][27][3,18,27]. This domain is important for YAP/TAZ regulation. Its absence causes cytoplasmic re-localisation and activity inhibition. YAP and TAZ are able to fixe Zonula occludens-2 protein (ZO-2) (and also ZO-1 for TAZ) and co-localise with it in the nucleus [28][29][28,29]. In addition, Vestigial-like 1-4 (VGLL1-4) contain Tondu domains (TDU), which have the capacity of binding TEAD. In so doing, VGLL4, for example, is able to inhibit YAP-TEAD1 interaction, and target TEAD1 for degradation [30]. Likewise, Runt-related transcription factor 3 (RUNX3) physically interacts with TEAD at its N-terminal region and, consequently, stops YAP from fixing to it and reduces its DNA-binding capacity as well as the downstream signalling [31].
YAP/TAZ-TEAD regulation is, therefore, most of the time achieved via the hippo kinase members, whether inhibiting their function or not.
1.2.2. Hippo Kinase Core and Upstream Partners Regulation
The Hippo kinases are also very well regulated, allowing them to sense the need to exert their inhibitory effect on YAP and TAZ. Among the major regulators of the Hippo pathway are mechanical cues such as cell density, cell-cell adhesions, and apico-basal polarity [3][32][3,32]. Proteins involved in the maintenance of these cellular events are, thus, implicated in Hippo regulation.
Merlin, coded by neurofibromatosis gene NF2, is an essential regulator of Hippo kinases. It is localised at tight and adherens junctions, when cells are at high density and induce activation of core kinases, which in turn inhibit YAP/TAZ effectors to limit cell proliferation [33][34][33,34]. Merlin induces YAP phosphorylation by LATS by several means: it (1) binds with LATS, transporting it to the cell membrane and stimulating a complex formation with MST and SAV and its further activation; (2) promotes the assembly of scaffold proteins to facilitate LATS-YAP interaction and phosphorylation. Actin disruption can activate this Merlin-dependent process [35]. Merlin is critical in liver homeostasis and stem cell niche regulation [36].
Another important protein in the maintenance of cell and tissue integrity and, thus, having a role in the regulation of the Hippo pathway, is Scribble, an adaptor protein, which is important in cell polarity. It is localised at the cell membrane and, like Merlin, is able to assemble a complex, here made of MST, LATS, and TAZ, which is necessary for LATS activation and the consequent phosphorylation cascade [37].
Furthermore, apical proteins such as Crumbs and the Angiomotin family (AMOTs), as well as cadherins-actin cytoskeleton linker proteins such as α-Catenin, are involved in YAP/TAZ regulation by managing their localisation through phosphorylation [38][39][40][41][38,39,40,41].
Hippo pathway regulation can also imply cell surface receptors such as G-protein coupled receptors (GPCRs) [32] and the Leukaemia Inhibitory Factor receptor (LIFR) [42][43][42,43], part of the JAK/STAT signalling pathway. In addition, AMOTp130 isoform can also act as substrate for LATS1/2. AMOTp130-LATS1/2 signalling is inhibited by serum LPA, through activation of Gαs-coupled GPCR signalling. In this context, the LATS1/2 Ser-175 phosphorylation of AMOTp130 can disrupt the latter’s interaction with F-actin and decrease stress fibres and focal adhesions. In so doing, LATS1/2 and AMOTp130SER175 repress endothelial cell migration in vitro and angiogenesis in zebrafish models [44].
YAP/TAZ activation proteins also exist, for example, the Protein phosphatase 2A (PP2A) complex STRIPAK, which exerts a negative control on Hippo kinases through their de-phosphorylation, thus inducing YAP/TAZ [45].
1.3. Hippo and Cancer
This pathway’s implication in cellular and tissular vital processes makes its dysregulation highly critical. Despite the numerous levels of regulation, the hippo pathway is found to be involved in various cancers. In Drosophila, mutations altering the function of Hippo kinases induce hyperproliferation and decreased apoptosis, leading to the appearance of tumours. This first demonstrated the tumour suppressor characteristics of Hippo kinases [3][12][3,12]. The liver tissue enlargement observed after mutation of Hippo kinases can be attributed to uncontrolled cell growth observed in cancers [5][8][5,8]. Furthermore, activation of YAP drosophila homologue Yki led to unrestrained growth of cells, which is one prerequisite for tumour formation [5].
Studies relate the role of YAP/TAZ hyperactivation in many cancers. The critical genes regulated by these transcriptional co-activators make it easy to go over to the dark side when disturbed, tending towards some cancer hallmarks such as sustaining proliferative signalling, resisting cell death, activating invasion and metastasis, and tumour-promoting inflammation, among others [46][47][71,72]. Though it has high functional potential, activating mutations of YAP gene are not described in cancer. Nonetheless, it is found to be localised on the 11q22 amplicon, amplified in various cancers, which could explain its hyperactivity [48][73]. Furthermore, inactive mutations or epigenetic regulations of Hippo kinases or close members are often observed in tumours [48][73].
Plouffe et al. show that YAP and its paralogue TAZ have distinct roles apart from their overlapping functions [49][74]. Hippo target genes induction can also differ depending on the effector involved. For example, CTGF and CYR61 are regulated by both YAP and TAZ, since serum-induced stimulation of these genes is decreased in YAP/TAZ-KO cells. Furthermore, YAP deletion seems to have a greater effect than TAZ deletion on CTGF and CYR61 expression, though to a lower extent for the latter. This was demonstrated in three different cell lines, including HeLa cervical cancer, MCF7 breast cancer, and HEK293A human embryonic kidney cells. YAP KO, TAZ KO, and YAP/TAZ KO caused the induction of LGR5, showing the role of both effectors in LGR5 repression. YAP and YAP/TAZ KO demonstrated the role of YAP in cell spreading, size, granularity, glucose uptake, proliferation, and migration, which decreased after its deletion compared to wild-type (WT) cells that resembled TAZ KO cells [49][74]. Interestingly, LATS1/2 KO cells gave the opposite of what was observed in YAP KO and YAP/TAZ KO cells. Nevertheless, this researchtudy was carried out on HEK293A human embryonic kidney cells in which, despite the higher TAZ mRNA expression than that of YAP, the highly dynamic regulation of TAZ made its protein less than twice that expressed by the YAP protein [49][74]. This could explain why YAP deletion affects more cell size and physiology than TAZ deletion. YAP different regulation of CTGF is, nonetheless, a solid fact, and the other YAP-dependent target genes identified were AMOTL2 and Fos-like antigen 1 (FOSL1).
Colorectal (CRC), NSCLC, and breast cancers, as well as hepatocellular carcinomas (HCC) and melanomas, for example, present high expression of YAP or TAZ. In CRC, circular RNA circPPP1R22A induces YAP activation, causing tumour growth and metastasis, which decreases in presence of peptide 17, an inhibitor of YAP [50][75].
In breast-cancer-induced bone marrow metastasis, hypoxic environments influence HIF-1α interaction with TAZ. Nuclear HIF-1α is associated with the epithelial-to-mesenchymal transition (EMT) process and interaction with Hippo effectors, and is negatively regulated by LATS1/2 [51][52][76,77]. YAP interaction with ZEB1, as discussed before, contributes to the expression of target gene having a role in poor survival of patients, therapy resistance, and increased metastasis in breast cancer [53][68].
Furthermore, breast cancer bone marrow metastasis can also be induced by crosstalk between ROR1-Her3 and Hippo-YAP, through inactivation of MST1 [54][78]. Indeed, the phosphorylation of HER3 at Tyr1307 by the ROR1 tyrosine kinase receptor induces the methylation of MST1 at Lys59 and its deactivation. MOB1a/b deletion causes breast [55][79] and lung tumours [56][80]. Furthermore, it has been demonstrated that breast cancer cells are able to promote YAP/TAZ expression and activity in cancer associated fibroblasts (CAFS), one of the components of the tumour micro-environment. This has, as a consequence, the remodelling and stiffening of extracellular matrix, related to CAFs’ pro-tumorigenic roles, as well as the angiogenesis improvement required for tumour growth [57][81].
YAP and TAZ overexpression in NSCLC are related to tumour development, progression, and a patient’s poor prognosis. Hyperactivation mutations of YAP are observed in this type of cancer, as is the downregulation of LATS2 in 60% of cases [58][82]. miR-135 is found to be highly expressed in NSCLC and is associated with poor survival outcomes. This microRNA is capable of increasing invasive and migration properties of cancer cells in vitro and metastasis in vivo, through the targeting of hippo kinase core members [59][83]. Moreover, YAP is able to activate EMT transcription factor Slug, which in turn inhibits the BMF pro-apoptotic factor. Cells enter a senescence-like dormant state and counter the drug-induced apoptosis [60][84]. RAF/MEK/ERK is found to contribute to NSCLC through YAP modulation, since the anti-ERK1/2 siRNA strategy discussed earlier led to a decrease in migration and an invasion of NSCLC cells, along with the decrease in YAP protein expression observed [61][65].
In the liver, inducible YAP expression as well as deletion of NF2, SAV1, or MST1/2 leads to hepatomegaly and, further, to liver tumours [8][9][36][62][8,9,36,85]. HCC is characterised by the high expression of miRNA-665 and miRNA-3910, which have inhibitory effects on hippo kinases such as MST1, leading to decreased apoptosis and increased cell proliferation, migration, invasion, and EMT [18][63][64][65][18,86,87,88]. On the contrary, miRNA-195, having a tumour suppressor role, is decreased in HCC, and this is associated with the low survival rate of patients [66][67][89,90]. YAP activation is an early event in HCC, with the PDZ-binding domain being crucial for the activation of the cell proliferation gene CTGF [12]. In this type of cancer, CREB can promote transcriptional activity of YAP [68][69][91,92]. In addition, MEK1-YAP1 interaction leads to increased cell proliferation and maintenance of transformed neoplastic phenotype [70][93].
In melanomas, YAP is sufficient and necessary for invasion of cells and appearance of spontaneous metastasis [71][94]. Target genes involved in this phenomenon are AXL, THBS1, and CYR61. In skin cancers, deletion of MST1/2, surprisingly, does not have any flagrant effect, though YAP activation is implicated in the keratinocytes’ hyper-proliferation and the squamous cell carcinoma that is overcome, following the deletion of the CTNNA1 encoding α-catenin [40]. Furthermore, methylation of the LATS1/2 promoter contributes to oral squamous cell carcinoma (OSCC) [72][73][95,96]. In addition, in pancreatic cancer, YAP1 cooperates with K-RAS and induces tumour survival and EMT, which is involved in cancer cell metastasis [74][97].
Β-catenin-YAP cooperation, as explained above, is important in the tumorigenicity of β-catenin active cells, namely SW480, SNU-C1, HCT116 colon cancer cells, AGS GC cells, and A549 lung cancer cells, among others [75][55]. Interestingly, conversely to what expected, neither β-catenin partner TCF2 nor YAP partner TEAD was implicated in this pro-tumorigenic effect [76][77][52,56]. TBX5 transcription factor is the one mediating the pro-proliferation signal by inducing the transcription of BCL2L1 and BIRC5, and YES1 is essential in this process.
The Hippo pathway also plays an important role in the resistance to therapy of many cancers, such as gliomas, retinoblastomas, endometrial, bladder cell, pancreatic, and ovarian cancers [78][79][80][81][82][83][98,99,100,101,102,103]. YAP-TEAD is involved in NSCLC cells’ escape to Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) treatment [60][84]. Indeed, resistant cells are found to have high expression and activity of YAP/TEAD.
Another dysregulation observed in cancers and linked to the Hippo pathway is EMT, which involves a loss of epithelial architecture. This affects Hippo membrane regulators such as Scribble, which gets delocalised, thus activating YAP/TAZ and contributing to tumorigenesis and CSCs [37]. This has also been demonstrated in breast cancer cells lacking E-cadherin, having a role in epithelium integrity, and where the Scribble delocalisation phenotype can be counteracted by Hippo kinases reactivation [42]. Indeed, a perturbed E-cadherin/α-Catenin complex leads to decreased YAP phosphorylation and induced cancer-related transcriptional activity.
The role of the Hippo pathway in tissue homeostasis is greatly responsible for tumour appearance and maintenance when dysregulated. Cancer is a complex disease, and YAP/TAZ oncogenic ability seems to be involved in diverse parts of this ailment, from tumour initiation to tumour-immune cells’ crosstalk, extracellular matrix remodelling, and dissemination [3][7][12][84][85][3,7,12,104,105]. Disbalance of Hippo-YAP/TAZ regulation is at the root of many cancers, amongst which is gastric cancer (GC), on which researchwers will focus in the next part of this researchview.
2. Hippo Pathway in Gastric Carcinogenesis
2.1. Hippo Pathway in Helicobacter-Mediated Gastric Carcinogenesis
GC is a major health concern, and the Hippo pathway is implicated, since the very beginning of gastric carcinogenesis induced by chronic infection with Helicobacter pylori (H. pylori) is classified as a type 1 carcinogen and the principal cause of GC [86][87][88][89][106,107,108,109].
MST1/2 and LATS1/2 Hippo kinases are found downregulated, and YAP/TAZ effectors upregulated, in GC [90][91][92][110,111,112] (Figure 23). Nuclear expression of YAP/TAZ, related to their activity, is associated with a poor prognosis in GC patients, particularly in those with intestinal-type GC for YAP [32][42][93][94][95][96][32,42,113,114,115,116]. YAP is found overactivated in gastric carcinogenesis, and H. pylori infection stimulates both Hippo downstream effectors YAP/TAZ, as demonstrated by transcriptomic analyses in gastric epithelial cells after H. pylori infection [86][106]. This effect is abrogated by infection with CagA-mutant strains, showing the role of H. pylori in YAP hyperactivation and, most particularly, of the bacterium’s CagA oncoprotein.
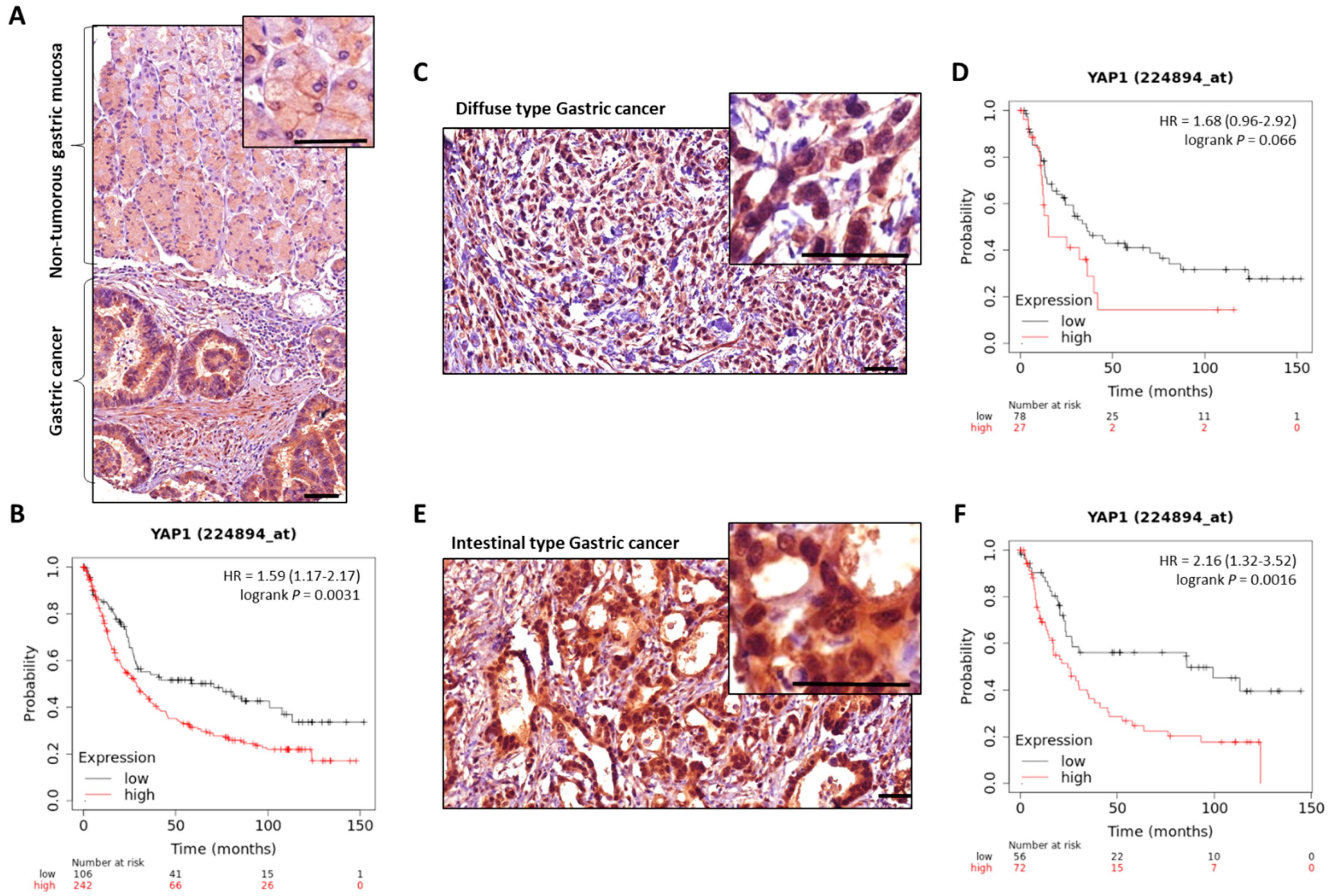
Figure 23. YAP expression in GC compared to non-tumorous gastric mucosa. Representative images of total-YAP immunostaining in GC patients’ tissues from our experiments. (A), non-tumorous gastric mucosa and adjacent GC from the same patient. (C,E) are representative images of diffuse and intestinal types GC cases, respectively; magnifications highlight YAP subcellular localisation, with nuclear accumulation in GC. Scale bars, 50 µm. KMplot™ analysis using KMplotter software (Kaplan-Meier plotter) [97][117] showing bad prognosis of patients with GC all subtypes included (B), diffuse (D), and intestinal (F) subtypes, when YAP is highly expressed.
Furthermore, YAP inhibition affected H. pylori-induced EMT, demonstrating the pathway’s contribution to H. pylori-related gastric carcinogenesis. Molina et al. have shown that H. pylori infection of gastric epithelial cells induces the expression of both YAP1 and LATS2 from the gastritis stage (early stage of the Correa’s cascade of H. pylori-induced gastric carcinogenesis) [43][86][43,106], which continues increasing at the intestinal metaplasia and GC stages [86][106]. YAP1-TEAD pro-proliferation and pro-survival-related genes were found to be increased after infection. Time-course infection with H. pylori showed that YAP1 is activated as early as 2h after the bacterial infection and causes an increase in target genes, including LATS2 acting as a regulatory feedback loop, thus controlling H. pylori-induced YAP/TAZ activation and cell growth from 5 to 24 h post-infection. An equilibrium, thus, exists for the maintenance of YAP1 proper balance as well as proper epithelial cell differentiation and survival in response to H. pylori infection, to limit epithelial cell identity loss [86][106].
Furthermore, nuclear YAP/TAZ-TEAD activity is induced following H. pylori infection of GC cells. TAZ is co-expressed and found co-localised with EMT-related transcriptional factor ZEB1 in cells having a mesenchymal phenotype, and at the invasive fronts of gastric tumours. TAZ-ZEB1 cooperation in H. pylori-induced GC is demonstrated by TAZ silencing, which decreases ZEB1 expression and EMT, as well as invasion and cancer stem cell (CSC) tumoursphere formation properties acquired by GC cells after H. pylori infection [87][107]. This could be related to direct TAZ interaction with the promoter of ZEB1, as described in RPE cells [98][69].
2.2. Hippo Pathway in GC and CSCs
CSCs’ presence in tumours is a major problem in GC, like in many other solid tumours [88][99][100][101][102][103][104][108,118,119,120,121,122,123]. CSCs correspond to a subpopulation of cells within tumours at the origin of tumour initiation, growth, dissemination, and resistance to current treatments. They have asymmetrical division properties, allowing their self-renewal and parallel proliferation and differentiation, at the base of tumour heterogeneity. Bessède et al. reported that, during the gastric carcinogenesis cascade, H. pylori infection induces the emergence of CD44+ cells carrying CSC properties, through the EMT process [105][124]. Giraud et al. recently demonstrated through transcriptomics analysis that CD44+ gastric CSCs have a Hippo-pathway-rich signature, with an overexpression of effectors and target genes such as AREG, CCND1, CDX2, CYR61, BIRC5, ID1, IGFBP3, JAG1, LATS2, SMAD7, and MYC [106][125]. Interestingly, TEAD1/4 were also found upregulated in CD44+ cells compared to CD44-non-CSCs, while TEAD inhibitors, VGLL4 and RUNX3, were downregulated. Furthermore, the researcheuthors also demonstrated that residual chemotherapy-treated cells, which are the most resistant ones, present higher expression of YAP1, TAZ, CYR61, and CTGF, all associated to high YAP/TAZ-TEAD activity, compared to non-treated cells.
Fujimoto et al. have shown that Protease-activated receptor 1 (PAR1) stimulation sustains cells with stem-like properties, and induces cell invasion and EMT through inhibition of LATS via Rho A GTPase [107][126]. PAR1 induces Rho A-induced inhibition of LATS1/2 phosphorylation, leading to activated YAP. Another receptor, Peroxisome proliferator-activated receptor δ (PPAR δ), is found to interact with YAP1, inducing the transcription and upregulation of SOX9, a gastrointestinal stem cell marker, in premalignant lesions [108][127]. SOX9 induces CSC properties in non-cancerous cells [109][128], promotes invasion and metastasis, and is overexpressed in patients at advanced stages of GC [11].
Gastric carcinogenesis process was also studied independently of H. pylori infection. Overexpression of hippo effectors, through LATS1/2 knockout (KO) in mice pyloric stem cells, generated transformation of mice normal gastric epithelium to low-grade intraepithelial neoplasia, followed by intramucosal carcinoma [110][129]. YAP/TAZ were found to initiate GC via MYC as a downstream mediator and direct transcriptional target of YAP [110][129]. Inhibition of MYC interfered with YAP/TAZ-induced carcinogenesis.
Another study showed LATS1 low expression in GC patients, and this was correlated with lymph node metastasis, poor prognosis, and tumour relapse [111][130]. LATS1 is found to decrease GC cell proliferation and invasion in vitro, and tumour growth and metastasis in vivo, by inhibiting YAP nuclear translocation and, thus, expression of pro-proliferative and pro-invasive target genes, among which are YAP, CTGF, PCNA, MMP-2, and MMP-9. TAZ overexpression, particularly in diffuse type GC, is associated with EMT profile and low survival of patients [43][106][112][43,125,131].
RUNX3 has been shown to be inactivated in about 60% GC. Subsequently, the tumour suppressor effect of this TEAD interactor on the YAP-TEAD complex is affected in GC, and hyperactivation is noted [31]. The RUNX3 mutation in GC impairs its activity towards a pro-YAP signalling phenotype. Likewise, RUNX2 is implicated in gastric carcinogenesis through the Hippo pathway. It is highly expressed in GC early stages and predicts poor prognosis [113][114][132,133]. Ectopic RUNX2 expression is implicated in GC invasion and metastasis process through binding to CXCR4 promoter, thus inducing the expression of CXCR4 [113][132]. It also induces tumoursphere formation and tumour initiation, showing an effect on the gastric CSC population [113][132]. CXCR4′s role is also described in CSC and metastasis in GC, as well as in pancreatic, breast, and colorectal cancer [115][116][117][118][119][134,135,136,137,138].
Zhou et al. show the role of methyltransferase 3 (METTL3), a major component of the m6A methyltransferase complex, in gastric carcinogenesis [120][139]. It is highly expressed in GC cases versus non-tumoral tissues. METTL3 overexpression and KD show its role in GC cell proliferation, migration, and invasion in vitro and tumour growth in vivo. Interestingly, METTL3 positively controls YAP1 expression, which could explain its effects [120][139].
Moreover, Angiomotin Like 1 (AMOTL1) has been shown to interact with YAP1 and promote its nuclear translocation and its activity, thus contributing to gastric carcinogenesis [121][140]. The hippo pathway is also involved in Fibroblast growth factor receptor type 2 (FGFR2) gastric oncogenesis, through indirect YAP1 activation via the MAPK-c-Jun pathway [122][141]. Overexpression of transcriptional core TEAD1/4 and YAP/TAZ targets CYR61 and CTGF is related to metastasis and bad prognosis. Furthermore, Tang et al. demonstrate a mechanism by which MST1/2 is turned off in some GC cases. Indeed, STRN3 was discovered as a PP2A regulatory subunit and was found to be able to recruit MST1/2, thus promoting its dephosphorylation and the consequent hippo effectors hyperactivation. dSTRIPAK forms part of the STRN3 molecule family, whose expression in induced in GC and associated with YAP activation and bad prognosis [123][142].
Additionally, microRNAs are also seen to intervene in the hippo pathway’s dysregulation in GC. MiR-125a-5p upregulates TAZ, TEAD2, and, therefore, their target genes, and stimulates cell survival, EMT, invasion, and tumour growth [64][87]. MiR-125a-5p is found in high levels at late stages of GC and co-expresses with TAZ and TEAD2. Similarly, miR-375 modulation is implicated in GC. Promoter methylation and histone deacetylation represses miR-375 expression in this disease, and this is associated with bad prognosis and lymph node metastasis [124][143]. Kang et al. show that miR-375 directly targets YAP1, TEAD, and CTGF, and that YAP1 re-expression partly stalls miR-375 tumour-suppressive effects. Furthermore, CTGF-KD demonstrates the same effects as YAP1-silencing and miR-375 ectopic expression on gastric tumours, demonstrating the role of Hippo effectors and their targets in GC. MiR-372 and MiR-373 are found to be implicated in GC cells AGS proliferation through inhibition of LATS2 gene expression [125][144].
Moreover, long noncoding RNAs (lncRNAs) have been demonstrated as having a role in hippo-pathway-induced gastric carcinogenesis. LncRNA RP11-323N12.5 is the most highly expressed lncRNA in GC, according to the TCGA database. RP11-323N12.5 is found to be correlated to YAP1 expression and to regulate it by acting on its promoter [126][145]. In so doing, it exerts pro-tumoral and pro-immunosuppressive functions. It is also regulated by YAP1/TAZ-TEAD transcription complex.
Besides, lncRNA acv3UTR is also found to be upregulated in GC. Ectopic expression of acv3UTR in GC cells demonstrated its role in cell growth promotion [127][146], through its negative effect on tumour suppressor miR-590-5p. Its expression correlates with that of YAP1. In addition, lncRNA LINC00649, enriched in GC, is found to induce cell proliferation, migration, and EMT in vitro, as well as tumorigenesis in vivo [128][147]. It is able to act on miR-16-5p, which is connected to YAP1 mRNA at the 3′UTR region. By doing so, it blocks the action of hippo kinases, which no longer phosphorylate YAP, and contributes to the expression of its pro-tumoral target genes [128][147]. On the contrary, Sun et al. identified lncRNA LATS2-AS1-001, which has an anti-tumoral effect in GC, through binding to the Enhancer of zeste homolog 2 (EZH2). This leads to LATS2 upregulation and YAP1 phosphorylation, thus decreasing cell viability, migration, and invasion. Apparently, this lncRNA is under-expressed in GC, which disables its effects [129][148].
Studies also show a relationship between cell metabolism and hippo-induced gastric carcinogenesis. Indeed, Liu et al. have shown the contribution of a nucleotide sugar transporter Solute carrier family, 35 B4 (SLC35B4), as having a crucial role in macromolecule glycosylation in GC [130][149]. The YAP1-TEAD complex directly activates SLC35B4, which induces cell proliferation in vitro and in vivo. It is more expressed in cancerous tissues compared to non-cancerous ones and is related to poor prognosis through YAP1′s action. In addition, Acylglycerol kinase (AGK), a lipid kinase involved in the production of physiological lysophosphatidic acid (LPA) and phosphatidic acid (PA), has been recognised for its pro-tumoral role when highly expressed [131][150]. It induces proliferation, invasion, and EMT phenotype in GC cells, effects that were abrogated by its KD. YAP1 plays a positive role on its regulation through binding to TEAD and to AGK’s promoter. In turn, in a positive feedback loop manner on YAP-TEAD, AGK inhibits hippo kinases, thus promoting YAP nuclear translocation and activity [131][150].
2.3. Hippo Pathway in GC Resistance to Therapy
The Hippo pathway has also been shown to have a role in therapy resistance in GC, like in many other cancers. There is, unfortunately, no targeted therapy in GC, except for Trastuzumab, which is approved for the treatment of HER2+ metastatic GCs. However, resistance mechanisms have been discovered that are also found at this level, rendering the search for other targets even more vital. Trastuzumab-resistant cells are found to have an overexpression of HER4, phosphorylated-HER4 (p-HER4), and vimentin (VIM), a mesenchymal phenotype marker, and a decrease in epithelial markers. Shi et al. demonstrate that in these cells, HER4 acts on its target YAP1 and induce the expression of genes involved in EMT and proliferation, leading to higher cells migration and growth as well as a decrease in HER2 and E-cadherin expression, leading to HER2-therapy resistance and contributing to mesenchymal phenotype acquisition [132][151]. Moreover, YAP stimulates GC cell proliferation, while its KD enhances cells’ sensitivity to cisplatin, showing the role of hippo effectors in GC chemoresistance [133][66]. In this rcontesearchxt, the researcheauthors showed that this effect passes through regulation of EGFR/AKT/Extracellular signal-regulated kinase ½ (ERK1/2) by YAP. TAZ overexpression, particularly in diffuse type GC, is associated with the EMT profile, resistance to chemotherapy, and low survival of patients [134][152].
Interestingly, studies show that YAP and TAZ are not always expressed concomitantly in GC cells [86][87][135][106,107,153]. TAZ is found to be more expressed in gastric signet ring cells carcinomas (SRCC), a poorly differentiated type of GC, usually attributed to Lauren-diffuse-type GC. This type of GC is of a particularly bad prognosis, and this could be related to its high TAZ expression, which is also found to be higher in other poorly differentiated tumours compared to well-differentiated ones such as the Lauren intestinal type. Tiffon et al. demonstrated that in the SRCC GC cell line MKN45 and poorly differentiated GC patient-derived xenograft cells, TAZ is overexpressed and overactivated, controlling the expression of TAZ-TEAD target genes, including CYR61 and AXL among others, and of the EMT main transcription factor ZEB1, which decrease in TAZ KD cells. TAZ is also related to GC cells’ EMT phenotype and invasiveness in this context [87][107]. Diffuse-type GC can also be characterised by low KRT17 intermediate filaments expression. This decreased KRT17 expression causes E-cadherin loss, EMT phenotype arousal, and metastasis in GC cells. Indeed, loss of intermediate filaments promotes cytoskeleton remodelling and reorganisation, which activates YAP and induces IL6 expression linked to increased metastasis [136][154].
The Hippo pathway and, most particularly, YAP and TAZ oncoproteins (Table 1), have a crucial role in gastric carcinogenesis, so therapeutic strategies involving their targeting could be of real efficiency in this poor prognosis, high relapse disease.
Table 1. Summary of YAP/TAZ expression, activation and regulation in GC.
| Expression Levels of YAP/TAZ | Regulation | Reference | ||
|---|---|---|---|---|
| Overexpression of YAP | Increase in pro-proliferation and pro-survival genes | [86] | [106] | |
| Upregulated through RUNX3 inactivation in GC | [31] | |||
| Induced by METTL3 found highly expressed in GC | [120] | [139] | ||
| Regulation by fixation of lncRNA | RP11-323N12.5 | on its promoter | [126] | [145] |
| Induced by HER4 and increases EMT, GC cells proliferation, and HER2-therapy resistance | [132] | [151] | ||
| Overactivation of YAP | Activated by PAR1 through inhibition of LATS Upregulation of stem-like properties |
[107] | [126] | |
| Interacts with AMOTL1 to promote its nuclear translocation and activity | [121] | [140] | ||
| Activation through MAPK-c-Jun pathway | [122] | [141] | ||
| Inhibition is decreased through PP2A- inhibition of MST1/2 | [123] | [142] | ||
| Overexpression of TAZ | Co-localisation with ZEB1 EMT transcription factor CSC tumorigenic properties |
[87] | [107] | |
| Upregulation by MiR-125a-5p, leading to stimulation of genes involved in cell survival, EMT, invasion, and tumour growth | [64] | [87] | ||
| Highly expressed in SRCC poorly undifferentiated GC | [135] | [153] | ||
| YAP/TAZ overexpression in CSCs and residual cells after chemotherapy-treatment | Overexpression of associated target genes | [106] | [125] |