Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Michael F. Olson.
Autosomal dominant lateral temporal epilepsy (ADLTE) is a genetic focal epilepsy associated with mutations in the
LGI1
,
RELN,
and
MICAL1
genes. A previous study linking ADLTE with two
MICAL1
mutations that resulted in the substitution of a highly conserved glycine residue for serine (G150S) or a frameshift mutation that swapped the last three C-terminal amino acids for 59 extra residues (A1065fs) concluded that the mutations increased enzymatic activity and promoted cell contraction.
- autosomal-dominant lateral temporal epilepsy
- LGI1
- RELN
- MICAL1
1. Clinical Characteristics and Genetic Transmission of ADLTE
Autosomal dominant lateral temporal epilepsy (ADLTE), also known as autosomal dominant epilepsy with auditory features, is a form of genetic focal epilepsy characterized by seizures affecting one hemisphere of the brain [1,2][1][2]. ADLTE onset is associated with mutations in the LGI1, RELN, or MICAL1 genes that code for the Leucine-rich Glioma-Inactivated 1 (LGI1), Reelin, and the molecule interacting with CasL 1 (MICAL1) proteins, respectively [3,4,5][3][4][5]. ADLTE is transmitted in an autosomal-dominant fashion, in which only one dominant acting allele is required for the phenotype to be manifested [6]. This disease typically presents in patients as auditory symptoms and aphasia, and inabilities with language comprehension, despite normal computed tomography (CT) and magnetic resonance imaging (MRI) results [7]. These presentations are usually required to establish a diagnosis, although clinicians may perform genetic testing for the previously mentioned genes if symptoms are inconsistent [7].
The typical developmental stages of onset are in adolescence and early adulthood, with an unknown but estimated very low prevalence within the population. In general, it has been estimated that epilepsies transmitted in a Mendelian manner, including ADLTE, account for a small percentage of all individuals living with the condition [8]. Much of what is known about ADLTE has been discovered through small-scale family studies with individuals either suspected or confirmed to have the disease, dating back to studies conducted in the mid-1990s [9]. Given the debilitating symptoms for those individuals living with ADLTE, more basic science and clinical research is required to understand the etiologies and molecular pathogenesis of the disease.
2. LGI1, Reelin and MICAL1 Genes in the Etiology of ADLTE
2.1. LGI1
The LGI1 gene is located on chromosome 10 band 10q23.33, and LGI1 is a secreted glycoprotein expressed in neurons [10]. LGI1 has been hypothesized to regulate the expression of Kv1 potassium channels through its interactions with membrane-anchoring proteins such as members of the “a disintegrin and metalloproteinase” (ADAM) family including ADAM22, and the postsynaptic membrane-associated guanylate kinase (MAGUK) protein family, thus ensuring normal neuronal signaling [11,12][11][12]. LGI1 forms a heterotetramer with ADAM22 in neuro, and this protein complex has been suggested to regulate receptors such as α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartic acid (NMDA) receptors [13,14][13][14]. In addition, Kv1 channels responsible for neuronal repolarization were found to be positively regulated by LGI1-ADAM22 complexes [15]. Two groups of LGI1 mutations have been proposed to affect these interactions, classified either as secretion-defective or secretion-competent [16]. In secretion-defective mutations, misfolded protein products are degraded by the endoplasmic reticulum and are therefore unable to bind to ADAM22 [13]. Disruptions in electrical currents may occur as a result. In secretion-competent mutations, the protein is secreted, but mutations render it incapable of binding ADAM22 [13]. However, there are other secretion-competent mutations that result in binding to ADAM22 but failure to form heterotetramers [17]. In any case, defective LGI1-ADAM22 complexes can disrupt Kv1-mediated currents, resulting in a failure to inactivate the neuron following depolarization [12,13][12][13] and consequent neuronal overexcitability, which ultimately may be responsible for the epilepsy symptoms observed in ADLTE (Figure 1a,b) [12,14][12][14].
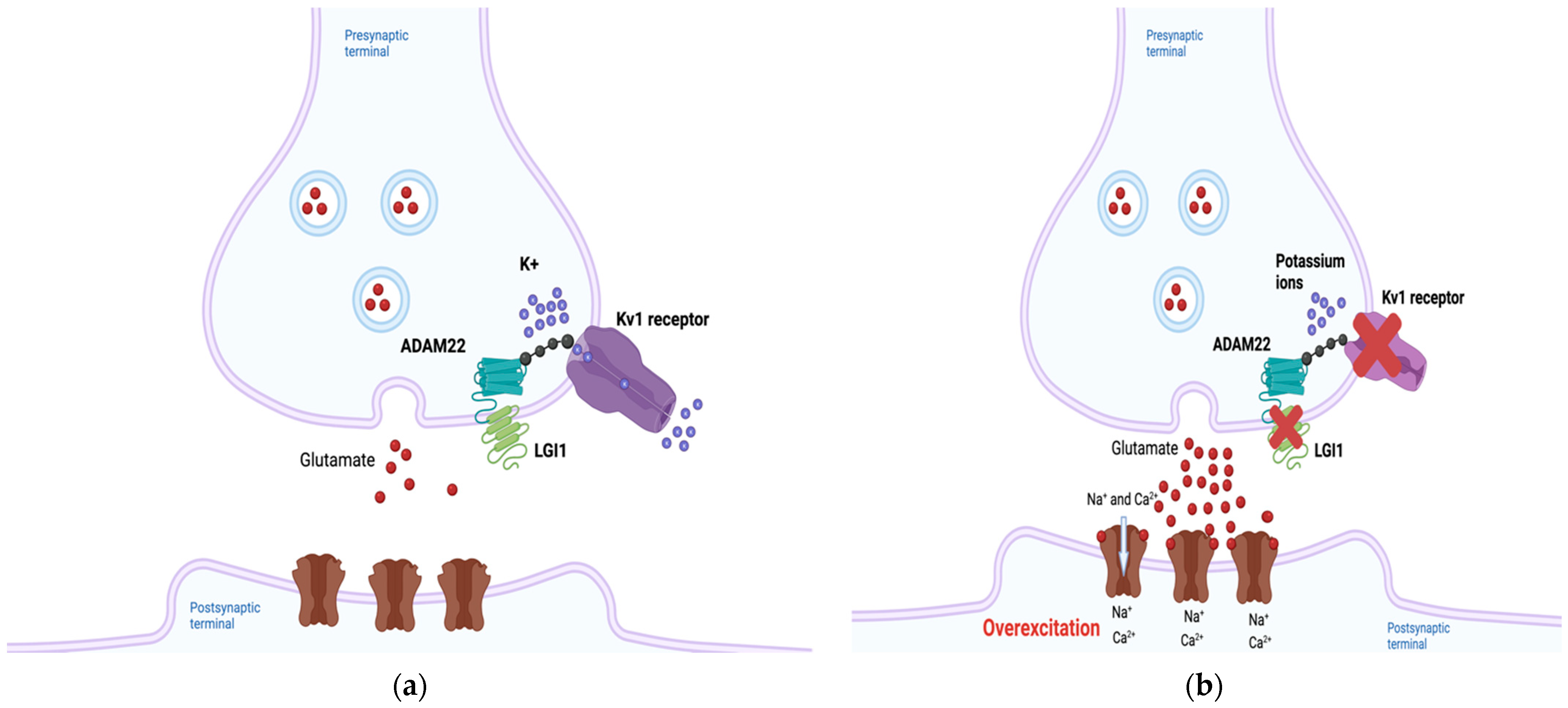
Figure 1. The predicted pathogenic mechanism of Autosomal Dominant Lateral Temporal Epilepsy (ADLTE)-associated LGI1 mutants. (a) Leucine-rich Glioma-Inactivated 1 (LGI1) normally regulates neuronal currents through its indirect association with potassium channels. (b) Through several different mechanisms, mutant LGI1 is unable to decrease Kv1 currents, inhibiting repolarization and subsequently increasing glutamate secretion, overexciting the postsynaptic neuron. Created with BioRender.com (accessed on 27 February 2022).
Mutant LGI1-mediated alterations in glutamatergic synapse development have also been proposed as a causative mechanism of ADLTE [18]. During the postnatal period, neurons downregulate the release of glutamate from presynaptic neurons and reduce expression of on postsynaptic neurons. LGI1 was shown to play a key role in this NMDA receptor downregulation [18]. Transgenic mice harboring a truncated LGI1 mutation (835delC) implicated in ADLTE showed increased presynaptic glutamate release and NMDA-receptor expression in the neurons of the hippocampal dentate gyrus compared to mice with the wild-type form of the protein [18]. The lower epileptic threshold of patients with ADLTE could result from the increased activation of excitation circuitry that remains throughout their lives [19].
Over 40 ADLTE-causing LGI1 mutations have been identified, with most mutations inhibiting the secretion of the protein into the extracellular matrix [17,20][17][20]. However, some studies have also found secretion-independent pathogenic mechanisms, leaving open the possibility of other mechanisms being responsible for the molecular pathogenesis [21]. The ADLTE-associated LGI1 mutation accounts for 67% of patients manifesting symptoms of the disease, and an estimated 30% of total ADLTE diagnoses are attributable to LGI1 mutations [1,22][1][22]. Current research on the involvement of LGI1 in ADLTE is largely focused on identifying and characterizing new mutations [23].
2.2. RELN
The RELN gene is located on chromosome 7 band q22.1, and the Reelin gene product is a secreted glycoprotein that contributes to the proper migration of cells in parts of the embryonic forebrain during embryogenesis, and axonal, dendritic and synapse development in the adult brain [24]. ADLTE patients with RELN mutations were found to have lower serum levels of the protein than those without the mutation [4]. Clarification of a potential relationship between the amount of Reelin and the pathogenesis of ADLTE came from a follow-up study that found that low levels of Reelin in the blood and extracellular matrix in the brain could lead to epilepsy [25], summarized in Figure 2. However, the precise mechanisms by which mutant Reelin might cause ADLTE have yet to be elucidated.
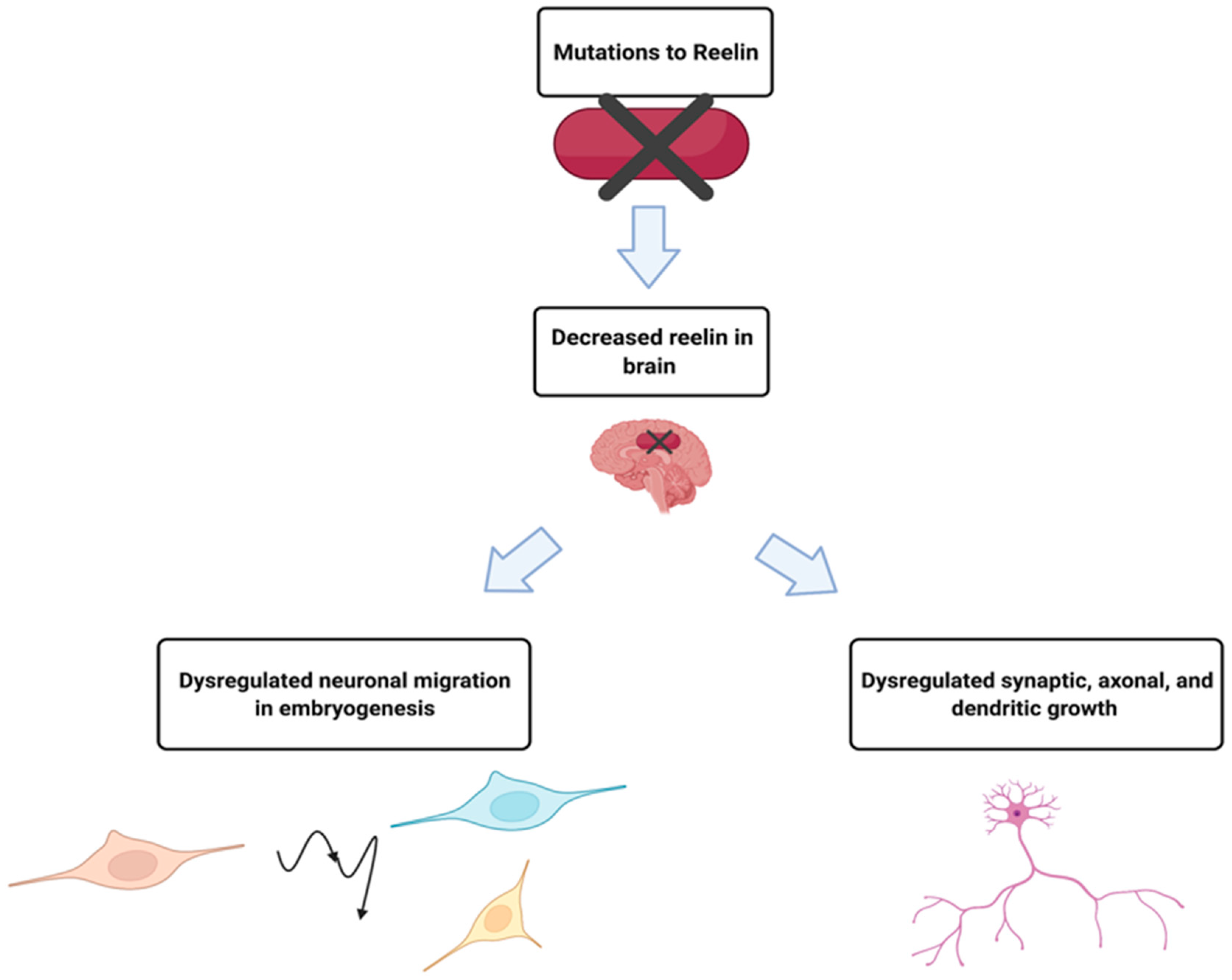
Figure 2. While much is unknown about the mechanism by which mutant Reelin leads to ADLTE, decreased serum levels in ADLTE patients suggested causal roles. It is hypothesized that Reelin is also decreased in the brain of these patients, affecting processes such as neuronal migration, synaptic potentiation, and dendritic growth, which could result in the development of ADLTE. Created with BioRender.com (accessed on 27 February 2022).
Pathogenic mutations in the RELN gene are thought to be causal in 17–18% of ADLTE diagnoses [1]. Since research has only recently implicated RELN in ADLTE, the penetrance of the mutations is unknown [1]. In a study that included 33 ADLTE-diagnosed patients, 60% of individuals showed a phenotype characteristic of ADLTE, consistent with the mutation having relatively high penetrance [4]. Additional research is needed to understand the pathogenic mechanism of mutant Reelin.
2.3. MICAL1
Isolated genetic case reports have suggested that unknown mutant genes at various chromosomal loci are linked to ADLTE-like symptoms and seizures [26,27][26][27]. An estimated 50% of ADLTE cases could be due to mutations of these uncharacterized genes [7]. Mutations of MICAL1, located on chromosome 6 band q21, were recently found to be linked to ADLTE in a cohort of Italian families [3,10][3][10]. In two families, researchers identified two different mutations to MICAL1: a glycine to serine substitution at the 150th amino acid (G150S), and a frameshift deletion mutation at the 1065th alanine (A1065fs) that resulted in the deletion of the last three C-terminal amino acids and the addition of 59 extra residues [3].
References
- Michelucci, R.; Pulitano, P.; Di Bonaventura, C.; Binelli, S.; Luisi, C.; Pasini, E.; Striano, S.; Striano, P.; Coppola, G.; La Neve, A.; et al. The Clinical Phenotype of Autosomal Dominant Lateral Temporal Lobe Epilepsy Related to Reelin Mutations. Epilepsy Behav. 2017, 68, 103–107.
- Stafstrom, C.E.; Carmant, L. Seizures and Epilepsy: An Overview for Neuroscientists. Cold Spring Harb. Perspect. Med. 2015, 5, a022426.
- Dazzo, E.; Rehberg, K.; Michelucci, R.; Passarelli, D.; Boniver, C.; Vianello Dri, V.; Striano, P.; Striano, S.; Pasterkamp, R.J.; Nobile, C. Mutations in MICAL-1 Cause Autosomal-Dominant Lateral Temporal Epilepsy: MICAL-1 Mutations in ADLTE. Ann. Neurol. 2018, 83, 483–493.
- Dazzo, E.; Fanciulli, M.; Serioli, E.; Minervini, G.; Pulitano, P.; Binelli, S.; Di Bonaventura, C.; Luisi, C.; Pasini, E.; Striano, S.; et al. Heterozygous Reelin Mutations Cause Autosomal-Dominant Lateral Temporal Epilepsy. Am. J. Hum. Genet. 2015, 96, 992–1000.
- Dazzo, E.; Santulli, L.; Posar, A.; Fattouch, J.; Conti, S.; Lodén-van Straaten, M.; Mijalkovic, J.; De Bortoli, M.; Rosa, M.; Millino, C.; et al. Autosomal Dominant Lateral Temporal Epilepsy (ADLTE): Novel Structural and Single-Nucleotide LGI1 Mutations in Families with Predominant Visual Auras. Epilepsy Res. 2015, 110, 132–138.
- Michelucci, R.; Pasini, E.; Malacrida, S.; Striano, P.; Bonaventura, C.D.; Pulitano, P.; Bisulli, F.; Egeo, G.; Santulli, L.; Sofia, V.; et al. Low Penetrance of Autosomal Dominant Lateral Temporal Epilepsy in Italian Families without LGI1 Mutations. Epilepsia 2013, 54, 1288–1297.
- Michelucci, R.; Nobile, C. Autosomal Dominant Epilepsy with Auditory Features. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Ottman, R.; Winawer, M.R.; Kalachikov, S.; Barker-Cummings, C.; Gilliam, T.C.; Pedley, T.A.; Hauser, W.A. LGI1 Mutations in Autosomal Dominant Partial Epilepsy with Auditory Features. Neurology 2004, 62, 1120–1126.
- Ottman, R.; Risch, N.; Hauser, W.A.; Pedley, T.A.; Lee, J.H.; Barker-Cummings, C.; Lustenberger, A.; Nagle, K.J.; Lee, K.S.; Scheuer, M.L.; et al. Localization of a Gene for Partial Epilepsy to Chromosome 10q. Nat. Genet. 1995, 10, 56–60.
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26.
- Seagar, M.; Russier, M.; Caillard, O.; Maulet, Y.; Fronzaroli-Molinieres, L.; De San Feliciano, M.; Boumedine-Guignon, N.; Rodriguez, L.; Zbili, M.; Usseglio, F.; et al. LGI1 Tunes Intrinsic Excitability by Regulating the Density of Axonal Kv1 Channels. Proc. Natl. Acad. Sci. USA 2017, 114, 7719–7724.
- Baudin, P.; Cousyn, L.; Navarro, V. The LGI1 Protein: Molecular Structure, Physiological Functions and Disruption-Related Seizures. Cell. Mol. Life Sci. 2022, 79, 16.
- Fukata, Y.; Hirano, Y.; Miyazaki, Y.; Yokoi, N.; Fukata, M. Trans-Synaptic LGI1–ADAM22–MAGUK in AMPA and NMDA Receptor Regulation. Neuropharmacology 2021, 194, 108628.
- Schulte, U.; Thumfart, J.-O.; Klöcker, N.; Sailer, C.A.; Bildl, W.; Biniossek, M.; Dehn, D.; Deller, T.; Eble, S.; Abbass, K.; et al. The Epilepsy-Linked Lgi1 Protein Assembles into Presynaptic Kv1 Channels and Inhibits Inactivation by Kvβ1. Neuron 2006, 49, 697–706.
- Fukata, Y.; Chen, X.; Chiken, S.; Hirano, Y.; Yamagata, A.; Inahashi, H.; Sanbo, M.; Sano, H.; Goto, T.; Hirabayashi, M.; et al. LGI1–ADAM22–MAGUK Configures Transsynaptic Nanoalignment for Synaptic Transmission and Epilepsy Prevention. Proc. Natl. Acad. Sci. USA. 2021, 118, e2022580118.
- Yamagata, A.; Miyazaki, Y.; Yokoi, N.; Shigematsu, H.; Sato, Y.; Goto-Ito, S.; Maeda, A.; Goto, T.; Sanbo, M.; Hirabayashi, M.; et al. Structural Basis of Epilepsy-Related Ligand–Receptor Complex LGI1–ADAM22. Nat. Commun. 2018, 9, 1546.
- Yamagata, A.; Fukai, S. Insights into the Mechanisms of Epilepsy from Structural Biology of LGI1–ADAM22. Cell. Mol. Life Sci. 2020, 77, 267–274.
- Zhou, Y.-D.; Lee, S.; Jin, Z.; Wright, M.; Smith, S.E.P.; Anderson, M.P. Arrested Maturation of Excitatory Synapses in Autosomal Dominant Lateral Temporal Lobe Epilepsy. Nat. Med. 2009, 15, 1208–1214.
- Anderson, M.P. Arrested Glutamatergic Synapse Development in Human Partial Epilepsy. Epilepsy Curr. 2010, 10, 153–158.
- Senechal, K.R.; Thaller, C.; Noebels, J.L. ADPEAF Mutations Reduce Levels of Secreted LGI1, a Putative Tumor Suppressor Protein Linked to Epilepsy. Hum. Mol. Genet. 2005, 14, 1613–1620.
- Dazzo, E.; Leonardi, E.; Belluzzi, E.; Malacrida, S.; Vitiello, L.; Greggio, E.; Tosatto, S.C.E.; Nobile, C. Secretion-Positive LGI1 Mutations Linked to Lateral Temporal Epilepsy Impair Binding to ADAM22 and ADAM23 Receptors. PLoS Genet. 2016, 12, e1006376.
- Rosanoff, M.J.; Ottman, R. Penetrance of LGI1 Mutations in Autosomal Dominant Partial Epilepsy with Auditory Features. Neurology 2008, 71, 567–571.
- Hu, P.; Wu, D.; Zang, Y.; Wang, Y.; Zhou, Y.; Qiao, F.; Teng, X.; Chen, J.; Li, Q.; Sun, J.; et al. A Novel LGI1 Mutation Causing Autosomal Dominant Lateral Temporal Lobe Epilepsy Confirmed by a Precise Knock-in Mouse Model. CNS Neurosci. Ther. 2022, 28, 237–246.
- D’Arcangelo, G. Reelin in the Years: Controlling Neuronal Migration and Maturation in the Mammalian Brain. Adv. Neurosci. 2014, 2014, 597395.
- Dazzo, E.; Nobile, C. Epilepsy-Causing Reelin Mutations Result in Impaired Secretion and Intracellular Degradation of Mutant Proteins. Hum. Mol. Genet. 2021, 31, 665–673.
- Fanciulli, M.; Pasini, E.; Malacrida, S.; Striano, P.; Striano, S.; Michelucci, R.; Ottman, R.; Nobile, C. Copy Number Variations and Susceptibility to Lateral Temporal Epilepsy: A Study of 21 Pedigrees. Epilepsia 2014, 55, 1651–1658.
- Bisulli, F.; Naldi, I.; Baldassari, S.; Magini, P.; Licchetta, L.; Castegnaro, G.; Fabbri, M.; Stipa, C.; Ferrari, S.; Seri, M.; et al. Autosomal Dominant Partial Epilepsy with Auditory Features: A New Locus on Chromosome 19q13.11-q13.31. Epilepsia 2014, 55, 841–848.
More