Definition: An armed antibody or antibody–drug conjugate (ADC) is a vectorized chemotherapy, which results from the grafting of a cytotoxic agent onto a monoclonal antibody via a judiciously constructed spacer arm. ADCs have made considerable progress in 10 years. While in 2009 only gemtuzumab ozogamicin (Mylotarg®) was used clinically, in 2020, 9 Food and Drug Administration (FDA)-approved ADCs are available, and more than 80 others are in active clinical studies. This review will focus on FDA-approved ADCs, their limitations including their toxicity and associated resistance mechanisms, as well as new emerging strategies to address these issues and attempt to widen their therapeutic window. Finally, we will discuss their combination with conventional chemotherapy or checkpoint inhibitors, to allow ADCs to get a little closer to the magic bullet imagined by Paul Ehrlich at the beginning of the 20th century.
- antibody–drug conjugate
- ADC
- bioconjugation
- linker
- payload
- cancer
- resistance
- combination therapies
1. Introduction/History
Antibody–drug conjugates (ADCs) have made considerable progress in 10 years. ADC is a vector-based chemotherapy that allows the selective delivery of a potent cytotoxic agent within a tumor. An ADC results from the generally stochastic grafting of a cytotoxic agent onto a monoclonal antibody (mAb) via a judiciously constructed spacer arm [1,2]. This is a complex mixture of immunoconjugates with different DLD (drug loading and distribution) and DAR (drug-to-antibody ratio, corresponding to the number of cytotoxics grafted onto the mAb) [3]. In 2009 gemtuzumab ozogamicin (Mylotarg®) was the only ADC approved by the Food and Drug Administration (FDA) and 12 other candidates were in clinical studies [4]. At present, 8 other ADCs have been approved and more than 80 others are in active clinical studies, including 6 in phase III or pivotal phase II (Table 1) [5]. More than 50 candidates have also been abandoned in the clinic mainly for toxicological reasons or because of a lack of efficiency. ADCs targeting solid tumors are currently making a satisfactory breakthrough in the clinic. Until November 2019, only Kadcyla® had an indication in solid tumors. With the late 2019 FDA-approval of both Padcev® and Enhertu®, and Trodelvy® in April 2020, there are currently four FDA-approved ADCs directed against solid tumors. The other five ADCs are indicated in hematological cancers and are generally considered to be easier to target with ADCs. In addition, ADCs in the advanced clinical phase are mainly directed against solid tumors (four against solid tumors; two directed against lymphomas).
Table 1. Antibody–drug conjugates (ADCs) approved by the Food and Drug Administration (FDA).
| Company | ADC (Cytotoxic) | Isotype and Target | Indication/Approval Date (Trade Name)/Clinical Status |
|---|---|---|---|
| Pfizer | gemtuzumab ozogamicin (CAL) | IgG4 CD33 | 2000–2010/2017 AML (Mylotarg®) |
| Seattle Genetics | brentuximab vedotin (AUR) | IgG1 CD30 | 2011 ALCL and Hodgkin lymphoma (Adcetris®) |
| Roche | trastuzumab emtansine (MAY) | IgG1 HER2+ | 2013 metastatic HER2+++ breast cancer (Kadcyla®) * |
| Pfizer | inotuzumab ozogamicin (CAL) | IgG4 CD22 | 2017 ALL and CLL (Besponsa®) |
| Roche | polatuzumab vedotin (AUR) | IgG1 CD79b | 2019 DLBCL (Polivy®) |
| Seattle Genetics | enfortumab vedotin (AUR) | IgG1 Nectin 4 | 2019 urothelial cancer (Padcev®) * |
| Daiichi Sankyo | trastuzumab deruxtecan (EXA) | IgG1 HER2+ | 2019 metastatic HER2+++ breast cancer (Enhertu®) * |
| Immunomedics | sacituzumab govitecan (IRI) | IgG1 TROP-2 | 2020, metastatic TNBC (Trodelvy®) * |
| GSK | belantamab mafodotin (AUR, MMAF) | IgG1afuc BCMA | 2020, multiple myeloma (Blenrep®) |
* ADC targeting solid tumor.
In 2009, calicheamycins, auristatins and maytansinoids were the main classes of cytotoxics used for ADC development. Ten years later, these same molecules are still used among other payloads optimized for better stability and hydrophilicity. New classes of cytotoxics have also been developed (PBDs, duocarmycins and camptothecin derivatives). In 10 years, considerable progress has been made in antibody engineering to allow more site-specific conjugation, to improve the homogeneity and the stability of the constructions and to bring 2nd and 3rd generation ADCs to the clinic in the hope to broaden the therapeutic index (ratio of the median lethal dose (LD50) to the median effective dose (ED50)) [1]. Several dozen bioconjugation technologies based on cysteine residues, non-natural amino acids or patterns introduced by molecular engineering have been proposed in preclinical studies [6]. Finally, more tumor specific antigenic targets and optimized release mechanisms of the cytotoxic agent within the tumor have led to the development of more performant ADCs [7–9].
This review will focus on FDA-approved ADCs as well as their limitation including their toxicity and associated resistance mechanisms. We will describe new emerging strategies to deal with these issues, including 3rd generation molecular constructions, the choice of alternative vectors, innovative delivery systems and combinations of ADCs with conventional chemotherapy or immune checkpoint inhibitors.
2. Design, Mechanism of Action and Therapeutic Indications of FDA-Approved First- and Second-Generation ADCs
The development of immunoconjugates in oncology has enabled the emergence of two key elements necessary to ensure the success of an ADC. The first concerns the need of a linker between the mAb and the payload. This mAb-linker-payload system was first designed with a cleavable linker (Figure 1) assumed to be stable under physiological conditions during plasma circulation, and quickly cleaved after tumor cell endocytosis, in order to selectively deliver the payload in the tumor and limit the appearance of undesirable side effects due to off-target toxicities. This type of linker is sensitive to lysosomal conditions (proteases, acidity and a reducing medium). The second key element for an ADC is correlated with the necessity to have a powerful cytotoxic agent grafted to the antibody [10].
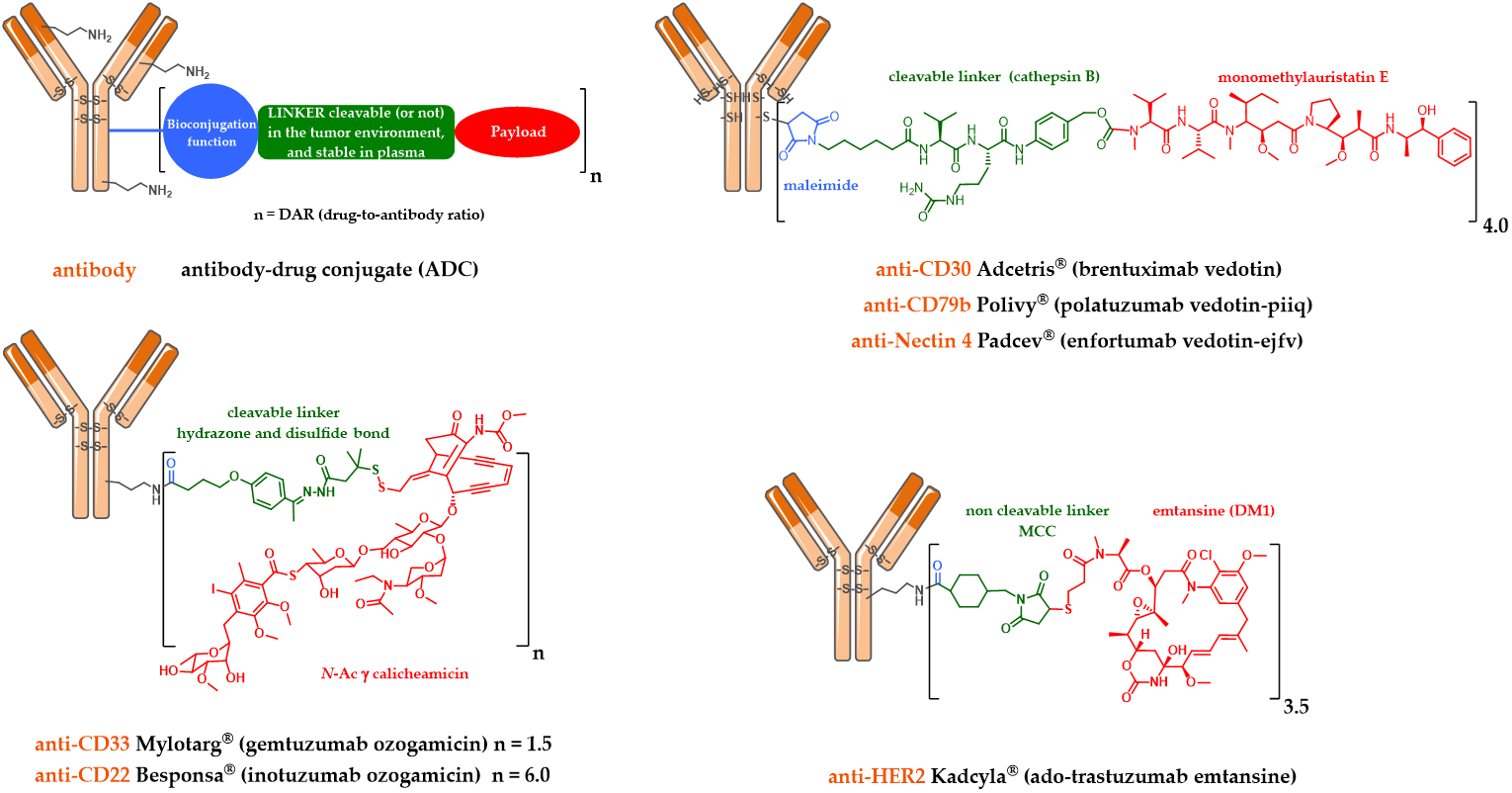
Figure 1. Schematic representation of the first and second generation FDA-approved ADCs: Mylotarg®, Adcetris®, Kadcyla®, Besponsa®, Polivy® and Padcev®.
2.1. Mylotarg®, Besponsa® and the First-Generation Cleavable Linker
Mylotarg® (Gemtuzumab ozogamicin) was approved by the FDA in 2000 for acute myeloid leukemia (AML) [11,12]. Mylotarg® results from the conjugation of calicheamycin, a powerful DNA-cleaving agent [13] with low nanomolar activity, onto gemtuzumab, a mutated anti-CD33 IgG4, via a cleavable linker including an hydrazone bond (Figure 1). This ADC, with an average DAR of 1.5, is a complex mixture with around 50% of unconjugated mAb [14]. After ADC internalization, the hydrazone bond can be hydrolyzed in the endosomal acidic environment to release a precursor of calicheamycin, which then undergoes a reduction by glutathione to the free active calicheamycin. The latter binds to the DNA minor groove and undergoes a Bergman cyclization, which generates a highly reactive diradical causing sequence-selective double-strand cuts (Scheme 1).
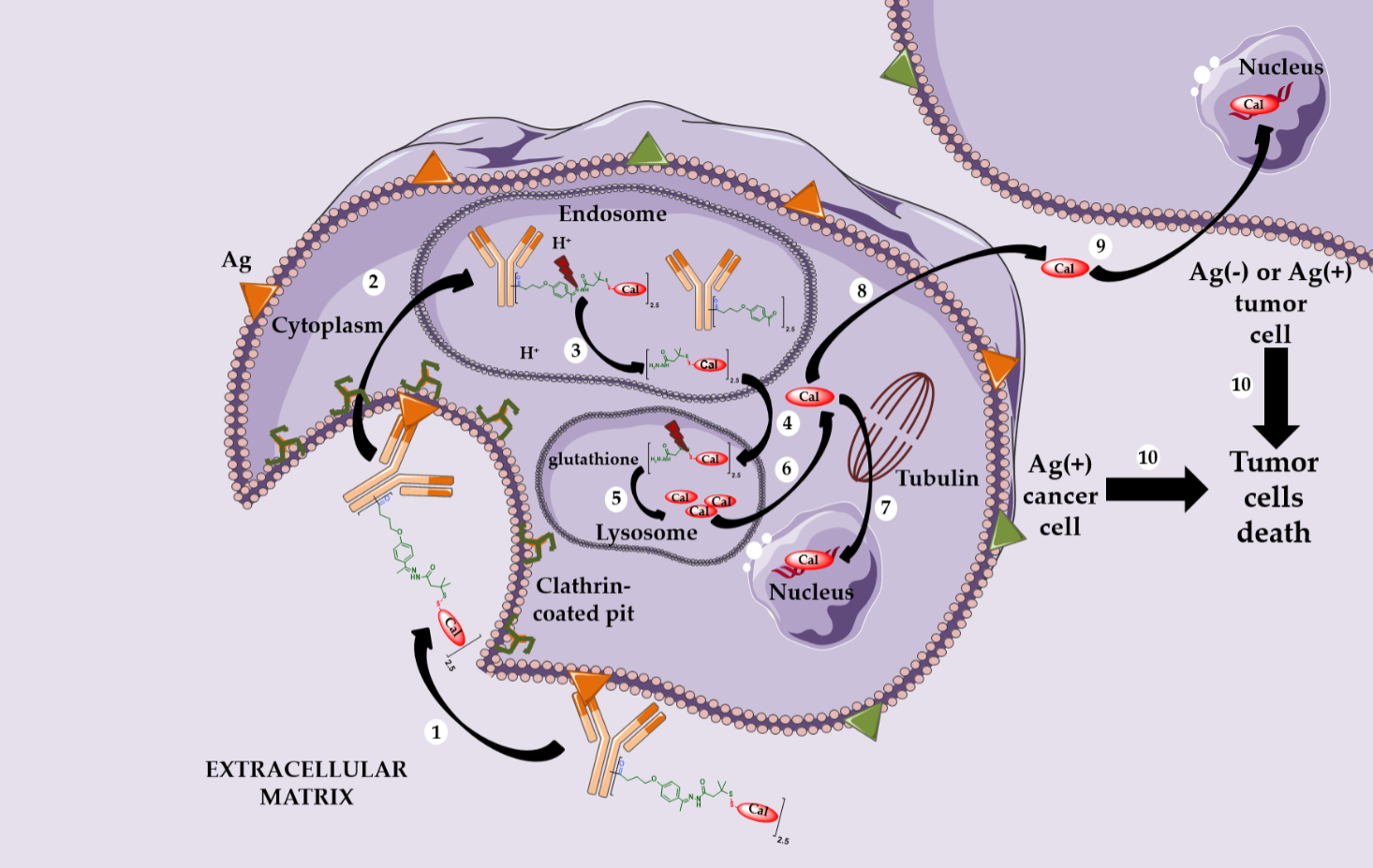
Theoretically, hydrazones should be stable in blood circulation at physiological pH and undergo selective hydrolysis after internalization in more acidic conditions (respectively pH 5.0–6.5 in endosomes and pH 4.5–5.0 in lysosomes).
However, the Mylotarg® linker exhibits a certain instability, leading to premature release of calicheamycin in plasma circulation [15], explaining its highly toxic profile and its subsequent voluntary withdrawal by Pfizer in 2010.
Mylotarg® benefited from the knowledge accumulated in the clinic in recent years to be reapproved by the FDA in 2017, used at lower doses, with a modified administration schedule and for a different patient population.
A similar linker was developed and used to graft calicheamycin onto inotuzumab, a mutated anti-CD22 IgG4, leading to Besponsa® (inotuzumab ozogamicin, Figure 1), approved by the FDA in 2017 against acute lymphoblastic leukemia (ALL) [16].
2.2. Kadcyla® and the Notion of Second Generation Non-Cleavable Linkers
Given these findings, alternative strategies for the design of linkers were necessary to continue the development of ADCs. Thus, Immunogen teams have focused on the use of linkers for the conjugation of maytansine derivatives and incorporating a delivery system using a glutathione-sensitive disulfide bond [17]. These innovative chemically labile linkers were intended to allow controlled release in the presence of glutathione (GSH), the cytoplasmic concentration of which in cancer cells is approximately 1000 times higher than in plasma.
In addition, the careful positioning of two methyl groups neighboring the disulfide bond enabled control of the release kinetics [18]. Thus, the high concentration of reducing molecules in the tumor should have guaranteed a selective release of the payload in the tumor environment but not in the circulation. This type of linker has not yet led to an ADC approved on the market.
However, a serendipitous finding allowed Immunogen to identify an unexpectedly effective ADC. The conjugation of DM1 onto the lysine residues of anti-HER2 IgG1 trastuzumab via a non-cleavable heterobifunctional thioether linker containing an N-hydroxysuccinimide ester (succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate or SMCC) led to Kadcyla® (T-DM1 or Ado-trastuzumab emtansine, Figure 1), which was approved by the FDA in 2013 [19]. This new ADC was observed in vitro to be very potent in a HER2-positive breast cancer model, the original structure being active only after internalization and complete enzymatic digestion of the ADC in the lysosome, to obtain the active metabolite lysine-MCC-DM1 (Scheme 2).
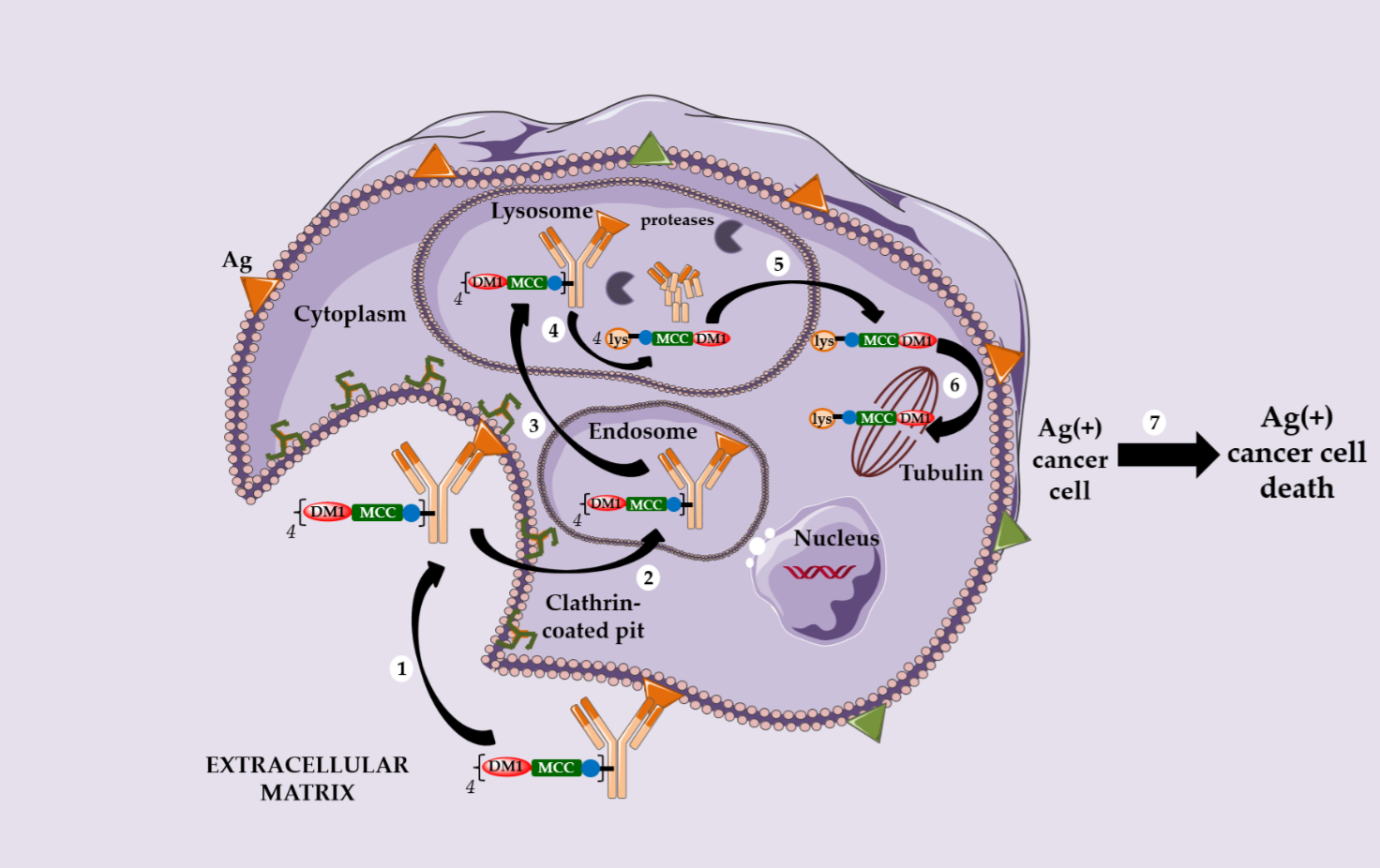
Following this discovery, several observations can be highlighted: (i) the metabolite lysine-MCC-DM1 retains the cytotoxic potential of free DM1, allowing the corresponding ADC to reach an in vitro activity in the picomolar range; (ii) this ADC exhibits no bystander killing effect due to the charged nature of its active metabolite at physiological pH; (iii) an ADC with a non cleavable linker can target only Ag-positive cells and (iv) this ADC has a limited toxicity on normal tissues and is more stable during circulation than an ADC with a cleavable linker.
2.3. Adcetris®, Polivy® and the Second-Generation Cleavable Linker
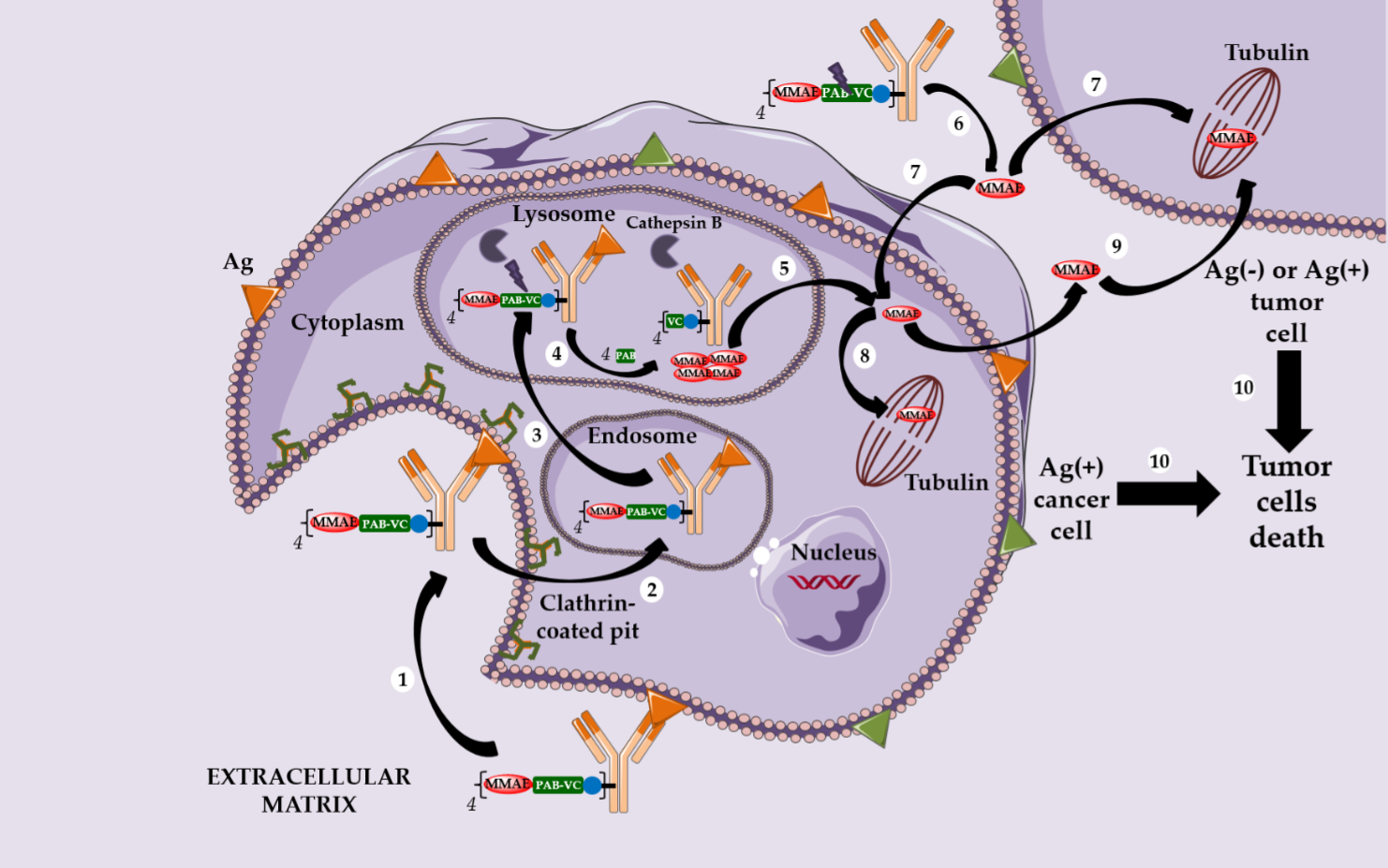
In parallel, Seattle Genetics has designed its own linker technology allowing the bioconjugation of dolastatin derivatives (such as monomethyl auristatin E or MMAE) onto the cysteine residues of an anti-CD30 IgG1 to produce Adcetris® (SGN-35 or brentuximab vedotin, Figure 1) [20–22]. After mild and partial reduction of the interchain disulfide bridges, the anti-CD30 mAb (cAC10) was bioconjugated to a cleavable heterobifunctional maleimide linker. This maleimidocaproyl-valine-citrulline-p-aminobenzyloxycarbonyl linker includes a valine-citrulline peptide trigger (ValCit) sensitive to lysosomal cathepsin B and a para-aminobenzyl alcohol (PAB) as a self-immolative spacer (SIS) allowing the release of MMAE after internalization in CD30-positive tumor cells.
Adcetris® was approved by the FDA in 2011 and for the treatment of anaplastic large cell lymphoma and Hodgkin’s lymphoma. After CD30-dependent ADC internalization, followed by degradation of the cleavable linker, the released MMAE can destroy the targeted cell and diffuse across the plasma membrane to reach and kill the neighboring cancer cells. This phenomenon is called the bystander killing effect [23,24] and allows the released MMAE to kill CD30-positive and CD30-negative tumor cells (Scheme 3) [25]. By corollary, the bystander killing effect explains the particular in vivo efficacy of Adcetris® in patients treated for heterogeneous lymphomas. Recently, Neri and his team demonstrated that the linker ValCit-PAB used in this ADC can also be cleaved before internalization [26], helping to dispel the dogma stipulating that an ADC must target an internalizing antigen [27] to be relevant.
Similarly, this second generation linker (mc-VC-PAB, Figure 1) was used to conjugate MMAE onto polatuzumab, an anti-CD79b IgG1, leading to Polivy® (polatuzumab vedotin-piiq, Figure 1), approved by the FDA in June 2019 in combination with bendamustine-based chemotherapy and rituximab, to treat adult patients with diffuse large B-cell lymphoma (DLBCL) [28,29].
We also found the same linker-drug (Figure 1) in enfortumab vedotin-ejfv, an ADC resulting from a stochastic bioconjugation on the cysteine residues of an IgG1 targeting Nectin 4, developed by Seattle Genetics, currently in a pivotal phase 2 clinical study [30]. On 16 July 2019, a BLA (Biologics License Application) for this ADC was submitted to the FDA for possible accelerated marketing for patients suffering from metastatic urothelial cancer and previously treated with anti-PD1/PDL1 antibodies. Enfortumab vedotin-ejfv was finally approved by the FDA as Padcev® in late December 2019.
3. Toxicity
The use of highly toxic ADCs has changed the paradigm of immunotherapy directed against tumor antigens, with limited side effects regarding the level of expression of the targeted antigen. Indeed, the two most used antibodies, rituximab directed against CD20 and trastuzumab directed against Her2, both have a toxicity profile allowing them to be combined with conventional chemotherapy agents without redundant toxicity. In the case of ADCs, the situation is different, because certain side effects are similar to some of those induced by conventional cytotoxic agents used in chemotherapy, while others are specific to the conjugates themselves. The occurrence of these side effects, sometimes severe, is partly explained by the uncontrolled release of the highly cytotoxic drug in the circulation, responsible for off-target toxicity. In addition, the IgG1 isotype of some of these ADCs can engage the Fc-gamma receptors (FcgR), which can trigger a target-independent, FcgR–dependent internalization in FcgR-positive cells resulting in toxic effects on these untargeted healthy cells. The development of these agents, either as monotherapy or in combination, has therefore proven to be complex.
Mylotarg® (gemtuzumab ozogamicin) can perfectly illustrate this statement. In 2004 a randomized study comparing a conventional treatment with an arm combining Mylotarg® was stopped prematurely due to an increased mortality rate in the latter. In 2010 Mylotarg® was withdrawn from most markets. However, study 0701 by the Acute Leukemia French Association (ALFA) showed that the fractionation of the administration into three doses improved both survival without events and overall survival without significant additional toxicity, in particular on the hepatic level [31]. The case of Mylotarg® is interesting for many reasons, it shows the difficulty of identifying a dose regimen to obtain a satisfactory therapeutic index, and the need not to abandon the development of an ADC after exploring only a single dose regimen.
In another case, Brentuximab vedotin (Adcetris®), directed against CD30, was approved for the treatment of certain forms of lymphoproliferative syndromes expressing CD30 including Hodgkin’s disease. The cytotoxic drug of this ADC is the vedotin (monomethyl auristatin E, MMAE), a powerful antitubulin agent. This ADC has a strong antitumor activity in monotherapy, in anaplastic large cell lymphomas (ALCL) and refractory Hodgkin’s diseases (NCT00848926) [32-34]. However, Adcetris®, as monotherapy, has been associated with potential severe peripheral neuropathies, neutropenia and thrombocytopenia, classic side effects of antitubulin agents.
Certain toxicities (e.g. bone marrow toxicity) observed with immunoconjugates are expected since the conjugates target either tubulin and the mitotic spindle (auristatins and maytansinoids) or DNA (calicheamycin and PBD). On the other hand, several side effects (including ocular toxicities or radionecrosis [35]), which are not observed with standard cytotoxic agents, have been reported. A better understanding and management of these unexpected toxicities will be essential for optimal use of these agents.
4. Mechanisms of resistance to ADCs
The mechanism of action of ADCs at the level of the tumor targeted cell comprises of several stages: binding to the antigen, internalization, release of the conjugate (mainly in the lysosome), release of the conjugate into the cytoplasm and then binding to the molecular target of the conjugate inducing cell death by apoptosis. Each of these steps can be involved in resistance as suggested by several preclinical works on cell lines or in animal models: (i) downregulation of the targeted antigen [36,37] and/or defects in binding, internalization, trafficking or recycling of the antibody, (ii) defective lysosomal degradation of the ADC or reduced expression of lysosomal transporters such as SLC46A3 [38], leading to lower release of the payload in the cytosol, (iii) alterations of tubulin or microtubule dynamics modulators [39] or (iv) reduced drug retention within the cell by upregulation of multidrug resistance transporters like MDR1 [40,41].
The clinical relevance of these various potential resistance mechanisms remains to be demonstrated. Indeed, it is complex to have access to tumor samples immediately before the initiation of treatment with ADC and then during the relapse following such treatments. Finally, in the context of therapeutic combinations, it can be complex to discern the mechanisms of resistance to ADCs from those of the other administered compounds. Despite this, the observations made on preclinical models raise interesting avenues for the analysis of resistance to ADCs in humans.
5. New strategies in Development: Third-Generation ADCs
Many ADCs, in clinical development or in clinical use, are based on a complete internalizing IgG format, targeting an Ag with an extremely high overexpression rate, and are conjugated to tubulin polymerization inhibitors using stochastic bioconjugation techniques [1]. In addition, cleavable linkers are known to be unstable during plasma circulation, while hydrophobic linkers are associated with a higher aggregation propensity. However, ADCs based on a complete IgG are associated with tumor penetration issues in stroma rich tumors [42,43] and are recycled by the neonatal Fc receptor (FcRn) leading to an undesirable distribution in the endothelium and the liver, responsible for undesirable side effects. Moreover, after internalization, ADCs effectiveness is based on favorable intracellular trafficking to reach the lysosome, where their degradation will allow controlled drug release. However, this strategy has several limiting factors. Firstly, an ADCs internalization capacity is intimately correlated with the high expression of surface Ag (for example, CD30 and HER2) [44], which explains why these ADCs using conventional tubulin polymerization inhibitors (for example, auristatins and maytansinoids) do not show cytotoxic activity on cells with low antigenic expression. Very powerful drugs (for example, pyrrolobenzodiazepine (PBD) dimers) have been developed to overcome these limitations, but the corresponding ADCs have a limited therapeutic index, particularly in solid tumors. Secondly, internalizing ADCs, including Kadcyla®, induce tumor resistance by several mechanisms. In fact, disturbances concerning internalization, trafficking or recycling of mAb, Ag shedding and defective lysosomal degradation of ADCs lead to a reduced drug release into the cytosol, thus compromising ADCs efficacy [36,37,45]. There is therefore today an important need to develop new technologies concerning bioconjugation techniques (leading to homogeneous ADCs), the vector format (antibodies or fragments), the linker (release mechanism) or the drug (new mechanism of action).
The development of first and especially second-generation ADCs has been associated with many dogmas, which many studies have considered as rules to follow in order to develop ADCs with better chances of success. Among these dogmas, we can cite targeting of an Ag not expressed in a ubiquitous manner, with a high overexpression level and internalizing, in particular to allow the intracellular release of the cytotoxic via a well-designed linker. The cleavable linker should be more stable than those sensitive to an acidic or reducing medium. The cytotoxic must have a potency at least like auristatins and maytansines to be used in ADC. Finally, the only format of interest for the antibody was IgG to guarantee a long half-time life and possibly the conservation of the effector activity from the parent mAb to the ADC.
Among the last ADC approvals by the FDA, GSK designed the ADC belantamab mafodotin (GSK2857916) by stochastically bioconjugating MMAF via a non-cleavable maleimide linker on an afucosylated IgG1 targeting BCMA (Figure 2). GSK2857916 has successfully finished a pivotal phase II clinical study against multiple myeloma [46], for patients whose disease has progressed despite prior treatment with an immunomodulatory agent, proteasome inhibitor and anti-CD38 antibody. Following a biologics license application (BLA) filled early in 2020, Blenrep® has just been approved by the FDA as well as by the EMA, as a first-in-class anti-BCMA therapy against multiple myeloma.
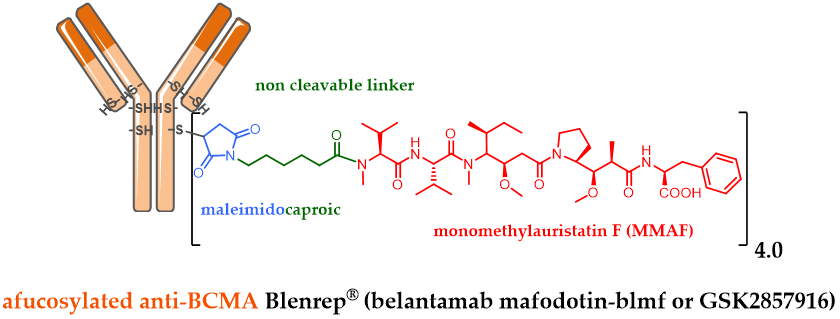
Figure 2. Formula of Blenrep® (belantamab mafodotin-blmf), an anti-BCMA antibody conjugated to MMAF via a non-cleavable linker.
Recent ADCs combine several innovations, among the target, format, release system, cytotoxic action mechanism and various bioconjugation techniques. In a disconcerting manner, despite the development of many technologies allowing a site-specific bioconjugation of a mAb to result in a more homogeneous ADC with an improved therapeutic index (10 in clinical study and more than 40 in preclinical), none of them has yet been validated by the approval of a homogeneous ADC.
Despite Ag specific targeting, ADCs are sometimes associated with high toxicity, specific or not to their target. This toxicity is linked to several mechanisms leading to the uncontrolled early release of the potent payload carried by the ADC outside the tumor. Unfortunately, in 2019, the ADCs area of research has not yet found the magic bullet that Paul Ehrlich dreamed of at the start of the 20th century. Given this observation, some companies have successfully turned to the development of original ADCs using less potent cytotoxic agents than MMAE or DM1, with new mechanisms of action to fight resistance to tubulin polymerization inhibitors. These ADCs also have release systems that are not necessarily specific for intracellular conditions and target original or unconventional targets.
For example, Immunomedics designed Sacituzumab govitecan (IMMU-132, Figure 3), an anti-TROP-2 [47] mAb conjugated to SN-38 (the active metabolite of irinotecan) via a cleavable maleimide linker (acidity) with a short pegylated unit [48]. The FDA-approval of this ADC was delayed and reached in April 2020, after a second BLA (biologics license applications) process was necessary to solve certain CMC issues following a successfully carried out phase III study. The accomplishment of this ADC is all the more impressive since it is indicated in refractory or resistant triple negative breast cancer (TNBC) against which there was no treatment until the FDA-approval of Trodelvy® in April 2020 [49]. Another interesting feature of this ADC: the optimization of the linker structure including a pegylated unit led to this ADC with a high DAR of 7.6 [47], without compromising its tolerance or efficiency. DAR 4 has long been considered as optimal, but this statement is now only true for the known approved ADCs with a second generation linker carrying DM1 or MMAE as the payload. IMMU-132 is a very interesting case study demonstrating that the optimal DAR of an ADC will depend on many parameters, mainly the hydrophilic nature of the linker and the grafted payload.
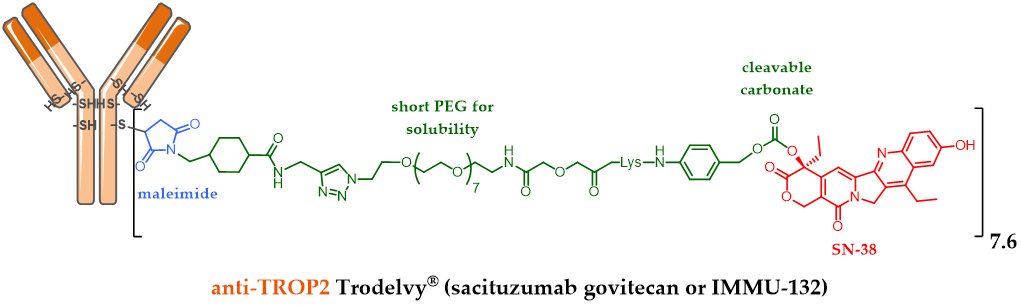
Figure 3. Trodelvy® (sacituzumab govitecan or IMMU-132) formula, ADC with a DAR 7.6, resulting from anti-TROP-2 antibody conjugation to SN-38 via an acid-sensitive cleavable linker.
Similarly, in order to conjugate an irinotecan derivative with a meticulously designed linker, the Japanese company Daiichi Sankyo developed DXd (exatecan or DX-8951). DXD is a cytotoxic agent 10-fold more active than SN-38 in vitro on cancer cells. DXD has a better safety profile with an optimized solubility, able to elicit a bystander killing effect [25] to kill neighboring cancer cells, which is an advantage in the heterogeneous tumor, but with a short half-life to avoid off-target toxicity. Bioconjugation of DXd onto the anti-HER2 trastuzumab cysteine residues via a maleimide linker sensitive to proteolysis made it possible to obtain the conjugate fam-trastuzumab deruxtecan-nxki (DS-8201a) with a homogeneous DAR of 7.7 (Figure 4) [50,51]. Despite its high DAR, Daiichi Sankyo’s DS-8201a was very well tolerated in rats and monkeys, and very stable in plasma (2.1% of DXD release after 21 days of incubation, in comparison to the release rate of T-DM1, which was 18.4% after only 4 days, despite the use of a non-cleavable linker). The use of a high DAR compound is remarkable as it contradicts the widely established principle that high DAR conjugates are unlikely to be good candidates due to poor pharmacokinetic profiles. These achievements were possible after testing many linkers. The chosen enzyme-sensitive linker encompasses a tumor-selective GGFC cleavable linker and an amino methylene SIS with a reduced hydrophobicity and a better plasma stability in comparison to the classical PAB SIS. Once DS-8201a is internalized, its linker is selectively cleaved by lysosomal proteases after the GGFG sequence to release a temporary DXD hydrolysate, which SIS amino-methylene is subsequently hydrolyzed to ammonia and formaldehyde to free DXD, triggering cell death.
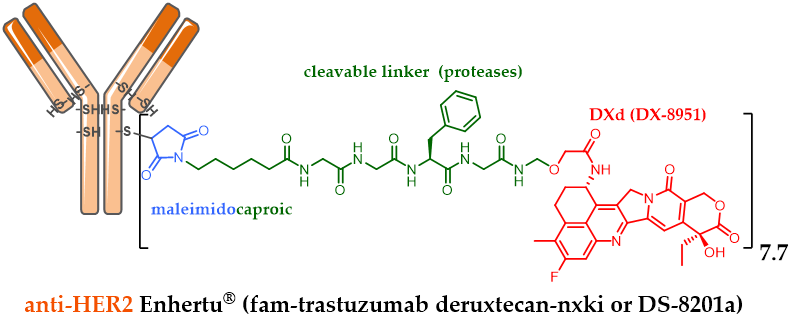
Figure 4. Enhertu® (fam-trastuzumab deruxtecan-nxki or DS-8201a) formula, ADC with a DAR 7.7, resulting from anti-HER2 trastuzumab conjugation to exatecan DX-8951 via a linker sensitive to proteolysis.
While Kadcyla® (T-DM1) is known to have in vitro efficacy only against HER2-positive cells with a high HER2 expression level, DS-8201a was effective in the pancreatic Capan-1 cell line with low HER2 expression and in the T-DM1 refractory JIMT-1 HER2-positive breast cancer cell line. With a DAR of 8 and a payload able to elicit the bystander killing effect, DS-8201a can effectively deliver DXD in a heterogeneous tumor in vivo and exhibit a high therapeutic effect [52]. DS-8201a was successfully tested against T-DM1 in a phase III clinical study in metastatic HER2-positive breast cancer last year. Enhertu® was finally approved by the FDA in late December 2019.
6. Indications of ADCs
6.1. cCombinations with Conventional Chemotherapy
Many studies are currently exploring combinations of Adcetris® or Kadcyla® with conventional chemotherapy. The objective may be here to replace an antitubulin agent with an ADC coupled with an antitubulin agent and likely to cause less toxicity, for example by replacing vincristine by Adcetris® in the treatment of certain lymphomas (NCT01777152). This could be particularly useful in fragile patients or those whose comorbidities preclude the use of conventional agents. Another objective may be to strengthen the activity of an established combination whose mechanism of action is different. As an example, Adcetris® is used with a combination of cisplatin, dexamethasone and cytarabine for the treatment of Hodgkin’s disease [53].
6.2. Adjuvant, Maintenance or consolidation Treatments
A growing number of patients are in remission from their cancers by a first line treatment, nevertheless without being in complete remission or cured. Several situations currently require adjuvant treatment (when the disease is not detectable) or maintenance (when the patients are in partial response). Kadcyla® has thus shown its superiority over trastuzumab, its unconjugated equivalent, in the randomized study KATHERINE (NCT01772472) as an adjuvant treatment in patients retaining residual breast or lymph node disease after neoadjuvant treatment with a decrease in 50% of the risk of local recurrence or death [54]. It is likely that other pathologies, in which maintenance treatment with naked mAbs have already proven their effectiveness, such as certain types of malignant lymphomas for example, can also benefit from the administration of ADC following a first line treatment.
6.3. Combinations of ADC and Immune Checkpoint Inhibitors
The value of combining cytotoxic chemotherapy with immune checkpoint inhibitors (ICPI) such as anti-PD1 and anti-PDL1 is currently the subject of numerous clinical studies [55]. In addition to the complementary mechanisms of action, the possibility of increasing the immunogenicity of tumors through immunogenic death induced by chemotherapy constitutes a strong argument for the association of certain cytotoxic agents with immunotherapies. The combination of ADC with ICPIs therefore appears to be a logical step, especially in patients who are already heavily pretreated and who want to avoid the systemic toxicity of chemotherapy. A phase 1/2 study of the combination of Adcetris® and nivolumab, an antibody directed against PD1, in patients with relapsed or refractory Hodgkin’s disease showed a response rate of 82%, including 61% complete responses [56]. These results were then confirmed in the Checkmate 205 study with responses in more than two thirds of the patients [57].
In general, ADCs have been shown to be effective in relapses and more recently in adjuvant situations in HER2-positive breast cancer. The positioning of these agents in the future will depend on several factors including the importance of the advantages brought compared to conventional chemotherapy (either in terms of toxicity or antitumor activity), the available therapeutic alternatives and the cost of patient care. ADCs are still a recent family of compounds. The approved agents are based on highly toxic payloads with mechanisms of action similar to those of conventional cytotoxic chemotherapy. The development of new ADCs based either on conventional agents such as sacituzumab govitecan, an antibody whose conjugate is the active metabolite of irinotecan [116] or on payloads with the original mechanisms of action, could also have a significant impact on the clinical use of ADC.
7. Conclusions and Perspectives
ADCs represent today a recent success of an old approach in chemotherapy targeting cancer. The classic internalizing ADCs currently used in clinics are designed to specifically deliver powerful cytotoxic agents to the targeted Ag (+) cancer cells, to eliminate only Ag (+) cancer cells (non-cleavable linker) or all tumor cells including both Ag (+) and Ag (−) cancer cells (cleavable and cytotoxic linker, rather hydrophobic). Over the past decade, ADCs have been improved by the choice of better cytotoxic agents, bioconjugation methodologies, better chosen targeted antigens and optimized antibody engineering.
However, despite their sophisticated design, ADCs are still associated with several limitations (for example, limited solid tumor penetration and toxicity) and the emergence of resistance mechanisms. To overcome these limitations, new antibody formats, new delivery systems, non-internalizing antigenic targets, new cytotoxic agents and site-specific bioconjugation methods have been studied to advance the development of ADCs. Unfortunately, many innovations have not yet been validated for use in clinical protocols, since the resulting ADCs are still in a preclinical or clinical study, and a small number have reached pivotal clinical phase II or phase III.
In conclusion, this field of research offers many encouraging prospects, especially when ADCs are combined with conventional chemotherapy or checkpoint inhibitors, in order to better potentiate their effects. In the last decade, the search for novel targets and the use of judiciously chosen payloads, with a well-designed release mechanism, have successfully opened the ADC field of applications. Hopefully, this research may allow forsaken drugs from the past to shine again as new conjugated drugs with better pharmacological profiles and efficacy, and allow ADCs to get a little closer to the magic bullet imagined by Paul Ehrlich at the beginning of the 20th century.
The article has been published on https://doi.org/10.3390/ph13090245
Author Contributions: Conceptualization, N.J., A.B., C.D. and C.D.-S.; investigation, N.J., A.B., C.D. and C.D.-S.; resources, N.J., A.B., C.D. and C.D.-S.; visualization, N.J., A.B., C.D. and C.D.-S.; validation, N.J., A.B., C.D. and C.D.-S.; writing—original draft preparation, N.J., A.B., C.D. and C.D.-S.; writing—review and editing, N.J., A.B., C.D. and C.D.-S.; funding acquisition, N.J. and C.D.-S; supervision, N.J. All authors have read and agreed to the published version of the manuscript.
Funding: Part of this work was supported by La Ligue contre le Cancer (comités 18, 35, 37, 41, 44, 53, 72, 79, 85), LabEx MAbImprove (ANR-10-LABX-53-01) and Région Centre Val de Loire (projects ARD 2020 Biomédicament and APR IR).
Conflicts of Interest: Alain Beck is an employee of Pierre Fabre, Charles Dumontet has received research funding from Roche and is a founder of Mablinks. All authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Rev. Drug Discov. 2017, 16, 315–337.
- Joubert, N.; Denevault-Sabourin, C.; Bryden, F.; Viaud-Massuard, M.C. Towards antibody-drug conjugates and prodrug strategies with extracellular stimuli-responsive drug delivery in the tumor microenvironment for cancer therapy. J. Med. Chem. 2017, 142, 393–415.
- Beck, A.; Terral, G.; Debaene, F.; Wagner-Rousset, E.; Marcoux, J.; Janin-Bussat, M.-C.; Colas, O.; Van Dorsselaer, A.; Cianférani, S. Cutting-edge mass spectrometry methods for the multi-level structural characterization of antibody-drug conjugates. Expert Rev. Proteomics 2016, 13, 157–183.
- Haeuw, J.F.; Caussanel, V.; Beck, A. Les immunoconjugués, anticorps «armés» pour combattre le cancer. Medecine/Sciences 2009, 25, 1046–1052.
- Beck, A.; Dumontet, C.; Joubert, N. Les immuno-conjugués en oncologie, les raisons du succès récent d’une approche ancienne. Médecine/Sciences 2019, 35, 1034–1042.
- Agarwal, P.; Bertozzi, C.R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Chem. 2015, 26, 176–192.
- Martin, C.; Kizlik-Masson, C.; Pèlegrin, A.; Watier, H.; Viaud-Massuard, M.-C.; Joubert, N. Antibody-drug conjugates: Design and development for therapy and imaging in and beyond cancer, LabEx MAbImprove industrial workshop, July 27–28, 2017, Tours, France. MAbs 2017, 10, 210–221.
- Denevault-Sabourin, C.; Bryden, F.; Viaud-Massuard, M.; Joubert, N. Antibody–Drug Conjugates: Empowering Antibodies for the Fight against Cancer. In Successful Drug Discovery; Wiley: Hoboken, NJ, USA, 2019; pp. 55–82.
- Beck, A.; Dumontet, C.; Joubert, N. Les immunoconjugués en oncologie, les nouvelles stratégies en développement. Médecine/Sciences 2019, 35, 1043–1053.
- Joubert, N.; Viaud-Massuard, M.-C. Antibody-Drug conjugates: Historical developments and mechanisms of action. In Optimizing Antibody-Drug Conjugates for Targeted Delivery of Therapeutics; Future Science Ltd.: Bielefeld, Germany, 2015; Volume 51, pp. 6–21, ISBN 9780124071919.
- Linenberger, M.L.; Hong, T.; Flowers, D.; Sievers, E.L.; Gooley, T.A.; Bennett, J.M.; Mark, S.; Leopold, L.H.; Appelbaum, F.R.; Bernstein, I.D.; et al. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2013, 98, 988–994.
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Chem. 2002, 13, 47–58.
- Ricart, A.D. Antibody-drug conjugates of calicheamicin derivative: Gemtuzumab ozogamicin and inotuzumab ozogamicin. Cancer Res. 2011, 17, 6417–6427.
- Beck, A.; D’Atri, V.; Ehkirch, A.; Fekete, S.; Hernandez-Alba, O.; Gahoual, R.; Leize-Wagner, E.; François, Y.; Guillarme, D.; Cianférani, S. Cutting-edge multi-level analytical and structural characterization of antibody-drug conjugates: Present and future. Expert Rev. Proteomics 2019, 16, 337–362.
- Trail, P.; Willner, D.; Lasch, S.; Henderson, A.; Hofstead, S.; Casazza, A.; Firestone, R.; Hellstrom, I.; Hellstrom, K. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215.
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. Engl. J. Med. 2016, 375, 740–753.
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Chem. Res. 2008, 41, 98–107.
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Chem. 2010, 21, 84–92.
- Erickson, H.K.; Lewis Phillips, G.D.; Leipold, D.D.; Provenzano, C.A.; Mai, E.; Johnson, H.A.; Gunter, B.; Audette, C.A.; Gupta, M.; Pinkas, J.; et al. The Effect of Different Linkers on Target Cell Catabolism and Pharmacokinetics/Pharmacodynamics of Trastuzumab Maytansinoid Conjugates. Cancer Ther. 2012, 11, 1133–1142.
- Sun, M.M.C.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.M.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody bisulfides. Chem. 2005, 16, 1282–1290.
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B. a; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Biotechnol. 2003, 21, 778–784.
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab vedotin (SGN-35). Cancer Res. 2011, 17, 6428–6436.
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. 2016, 76, 2710–2719.
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blattler, W.A.; Goldmacher, V.S. Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen. Cancer Res. 2006, 66, 3214–3221.
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046.
- Dal Corso, A.; Cazzamalli, S.; Gébleux, R.; Mattarella, M.; Neri, D. Protease-Cleavable Linkers Modulate the Anticancer Activity of Noninternalizing Antibody-Drug Conjugates. Chem. 2017, 28, 1826–1833.
- Teicher, B. Antibody-Drug Conjugate Targets. Cancer Drug Targets 2009, 9, 982–1004.
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; McCord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H.; et al. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586.
- An Anti-CD79B Antibody-Drug Conjugate Is Active in Non-Hodgkin Lymphoma. Cancer Discov. 2015, 5, 576–576.
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 2016, 76, 3003–3013.
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516.
- A phase 2 study of brentuximab vedotin in patients with relapsed or refractory CD30-positive non-Hodgkin lymphomas: Interim results in patients with DLBCL and other B-cell lymphomas. Adv. Hematol. Oncol. 2014, 12, 3–4.
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. Engl. J. Med. 2018, 378, 331–344.
- Younes, A.; Connors, J.M.; Park, S.I.; Fanale, M.; O’Meara, M.M.; Hunder, N.N.; Huebner, D.; Ansell, S.M. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: A phase 1, open-label, dose-escalation study. Lancet Oncol. 2013, 14, 1348–1356.
- Carlson, J.A.; Nooruddin, Z.; Rusthoven, C.; Elias, A.; Borges, V.F.; Diamond, J.R.; Kavanagh, B.; Kabos, P. Trastuzumab emtansine and stereotactic radiosurgery: An unexpected increase in clinically significant brain edema. Oncol. 2014, 16, 1006–1009.
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor Cells Chronically Treated with a Trastuzumab-Maytansinoid Antibody-Drug Conjugate Develop Varied Resistance Mechanisms but Respond to Alternate Treatments. Cancer Ther. 2015, 14, 952–963.
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Cancer Ther. 2015, 14, 1376–1384.
- Hamblett, K.J.; Jacob, A.P.; Gurgel, J.L.; Tometsko, M.E.; Rock, B.M.; Patel, S.K.; Milburn, R.R.; Siu, S.; Ragan, S.P.; Rock, D.A.; et al. SLC46A3 Is Required to Transport Catabolites of Noncleavable Antibody Maytansine Conjugates from the Lysosome to the Cytoplasm. Cancer Res. 2015, 75, 5329–5340.
- Sauveur, J.; Matera, E.-L.; Chettab, K.; Valet, P.; Guitton, J.; Savina, A.; Dumontet, C. Esophageal cancer cells resistant to T-DM1 display alterations in cell adhesion and the prostaglandin pathway. Oncotarget 2018, 9, doi:10.18632/oncotarget.24975.
- Yu, S.F.; Zheng, B.; Go, M.; Lau, J.; Spencer, S.; Raab, H.; Soriano, R.; Jhunjhunwala, S.; Cohen, R.; Caruso, M.; et al. A novel anti-CD22 anthracycline-based antibody-drug conjugate (ADC) that overcomes resistance to auristatin-based ADCs. Cancer Res. 2015, 21, 3298–3306.
- Chang, C.-H.; Wang, Y.; Zalath, M.; Liu, D.; Cardillo, T.M.; Goldenberg, D.M. Combining ABCG2 Inhibitors with IMMU-132, an Anti-Trop-2 Antibody Conjugate of SN-38, Overcomes Resistance to SN-38 in Breast and Gastric Cancers. Cancer Ther. 2016, 15, 1910–1919.
- Yasunaga, M.; Manabe, S.; Matsumura, Y. New concept of cytotoxic immunoconjugate therapy targeting cancer-induced fibrin clots. Cancer Sci. 2011, 102, 1396–1402.
- Yasunaga, M.; Manabe, S.; Tarin, D.; Matsumura, Y. Cancer-Stroma Targeting Therapy by Cytotoxic Immunoconjugate Bound to the Collagen 4 Network in the Tumor Tissue. Chem. 2011, 22, 1776–1783.
- Lambert, J.M.; Morris, C.Q. Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Ther. 2017, 34, 1015–1035.
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 3378.
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221.
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC)*. Oncotarget 2015, 6, 22496–22512.
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Arrojo, R.; Liu, D.; Rossi, E.A.; Chang, C.H.; Goldenberg, D.M. Sacituzumab govitecan (IMMU-132), an Anti-Trop-2/SN-38 antibody-drug conjugate: Characterization and efficacy in pancreatic, gastric, and other cancers. Chem. 2015, 26, 919–931.
- Govindan, S.V.; Starodub, A.N.; Juric, D.; Abramson, V.; Sharkey, R.M.; Wegener, W.A.; Tolaney, S.M.; Kalinsky, K.; O’Shaughnessy, J.; Maliakal, P.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. Clin. Oncol. 2018, 2017, 2141–2148.
- Nakada, T.; Masuda, T.; Naito, H.; Yoshida, M.; Ashida, S.; Morita, K.; Miyazaki, H.; Kasuya, Y.; Ogitani, Y.; Yamaguchi, J.; et al. Novel antibody drug conjugates containing exatecan derivative-based cytotoxic payloads. Med. Chem. Lett. 2016, 26, 1542–1545.
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Cancer Res. 2016, 22, 5097–5108.
- Nakada, T.; Sugihara, K.; Jikoh, T.; Abe, Y.; Agatsuma, T. Drug Discovery: Recent Progress and the Future The Latest Research and Development into the Antibody—Drug for HER2 Cancer Therapy. Pharm. Bull. 2019, 67, 173–185.
- Hagenbeek, A.; Mooij, H.; Zijlstra, J.; Lugtenburg, P.; van Imhoff, G.; Nijland, M.; Tonino, S.; Hutchings, M.; Spiering, M.; Liu, R.; et al. Phase I dose-escalation study of brentuximab-vedotin combined with dexamethasone, high-dose cytarabine and cisplatin, as salvage treatment in relapsed/refractory classical Hodgkin lymphoma: The HOVON/LLPC Transplant BRaVE study. Haematologica 2019, 104, e151–e153.
- von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. Engl. J. Med. 2019, 380, 617–628.
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Rev. Drug Discov. 2015, 14, 561–584.
- Herrera, A.F.; Moskowitz, A.J.; Bartlett, N.L.; Vose, J.M.; Ramchandren, R.; Feldman, T.A.; LaCasce, A.S.; Ansell, S.M.; Moskowitz, C.H.; Fenton, K.; et al. Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2018, 131, 1183–1194.
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. Clin. Oncol. 2018, 36, 1428–1439.
- Faltas, B.; Goldenberg, D.M.; Ocean, A.J.; Govindan, S.V.; Wilhelm, F.; Sharkey, R.M.; Hajdenberg, J.; Hodes, G.; Nanus, D.M.; Tagawa, S.T. Sacituzumab Govitecan, a Novel Antibody–Drug Conjugate, in Patients With Metastatic Platinum-Resistant Urothelial Carcinoma. Genitourin. Cancer 2016, 14, e75–e79.