The liver is one of the richest organs in mitochondria, serving as a hub for key metabolic pathways such as β-oxidation, the tricarboxylic acid (TCA) cycle, ketogenesis, respiratory activity, and adenosine triphosphate (ATP) synthesis, all of which provide metabolic energy for the entire body. Mitochondrial dysfunction has been linked to subcellular organelle dysfunction in liver diseases, particularly fatty liver disease. Acute fatty liver of pregnancy (AFLP) is a life-threatening liver disorder unique to pregnancy, which can result in serious maternal and fetal complications, including death. Pregnant mothers with this disease require early detection, prompt delivery, and supportive maternal care. AFLP was considered a mysterious illness and though its pathogenesis has not been fully elucidated, molecular research over the past two decades has linked AFLP to mitochondrial dysfunction and defects in fetal fatty-acid oxidation (FAO). Due to deficient placental and fetal FAO, harmful 3-hydroxy fatty acid metabolites accumulate in the maternal circulation, causing oxidative stress and microvesicular fatty infiltration of the liver, resulting in AFLP.
- liver
- mitochondrial dysfunction
- acute fatty liver of pregnancy
1. Acute Fatty Liver of Pregnancy (AFLP)
2. Mitochondrial Dysfunction and Pathogenesis of AFLP
2.1. Mitochondrial Dynamics and Biogenesis
Mitochondria are dynamic organelles that change their structure and shape in response to the energy demand and supply through fusion and fission processes [16,17][5][6]. Mitochondrial fission and fusion play important roles in maintaining the function of mitochondria under conditions of metabolic or environmental distress. Fusion is a process that helps mitigate cellular stress by mixing the contents of partially damaged mitochondria to promote complementation. Fission is needed to create new mitochondria, but it also contributes to quality control by enabling the removal of damaged mitochondria. Disruptions of these processes have been implicated in disease. Mitochondrial fission occurs when oxidative stress damages mitochondria, resulting in the separation of damaged mitochondria from healthy mitochondria [18,19][7][8]. Mitochondrial fusion-fission balance is disrupted by intracellular and external stress, resulting in mitochondrial fragmentation [20][9]. Mitochondrial dysfunction is associated with excessive fission, characterized by increased levels of the fission protein dynamin-related protein 1 (Drp1). The dysregulation of proteins involved in mitochondrial fission has an important impact on mitochondrial morphology and function [21,22][10][11]. In mammalian cells, mitofusin-1 (Mfn1) and mitofusin-2 (Mfn2) are the primary mediators of mitochondrial fusion and are responsible for the fusion process of the outer mitochondrial membrane (OMM) [23][12]. The protein Optic atrophy 1 (Opa1) regulates the fusion of the inner mitochondrial membrane (IMM), which is required for maintaining the balance between mitochondrial fusion and fission [24,25][13][14]. In clinical conditions involving placental dysfunction, changes in the expression of OPA1, SIRT3, and MFN1 have been identified [26,27][15][16]. PGC-1α (peroxisome proliferator-activated receptor gamma co-activator 1) and nuclear respiratory factor 1 (NRF1), which regulate expression of mtDNA and nuclear DNA genes encoding subunits of the MRC complexes and mtDNA replication and transcription, respectively, control mitochondrial biogenesis [28,29][17][18]. Because mitochondria are essential for metabolism and energy production [30,31,32][19][20][21] as well as regulating signaling pathways that mediate these processes [33[22][23],34], changes in mitochondrial dynamics can play an important role in the onset and progression of liver disease [35][24].2.2. Mitochondrial Fatty-Acid Oxidation (FAO)
2.2.1. Fatty Acid Transport to Mitochondria
Figure 1 provides a schematic representation of FA transport to mitochondria. Free fatty acids (FFAs) in the liver are derived from plasma FFAs produced by adipose tissue and chylomicrons, or they are synthesized de novo in the liver. These FFAs are either oxidized in mitochondria or esterified as triglycerides in hepatocytes, where they accumulate as fat droplets or are packaged with apolipoprotein B, cholesterol esters, and phospholipids to be secreted as very-low-density lipoproteins (VLDLs) [32,41][21][25].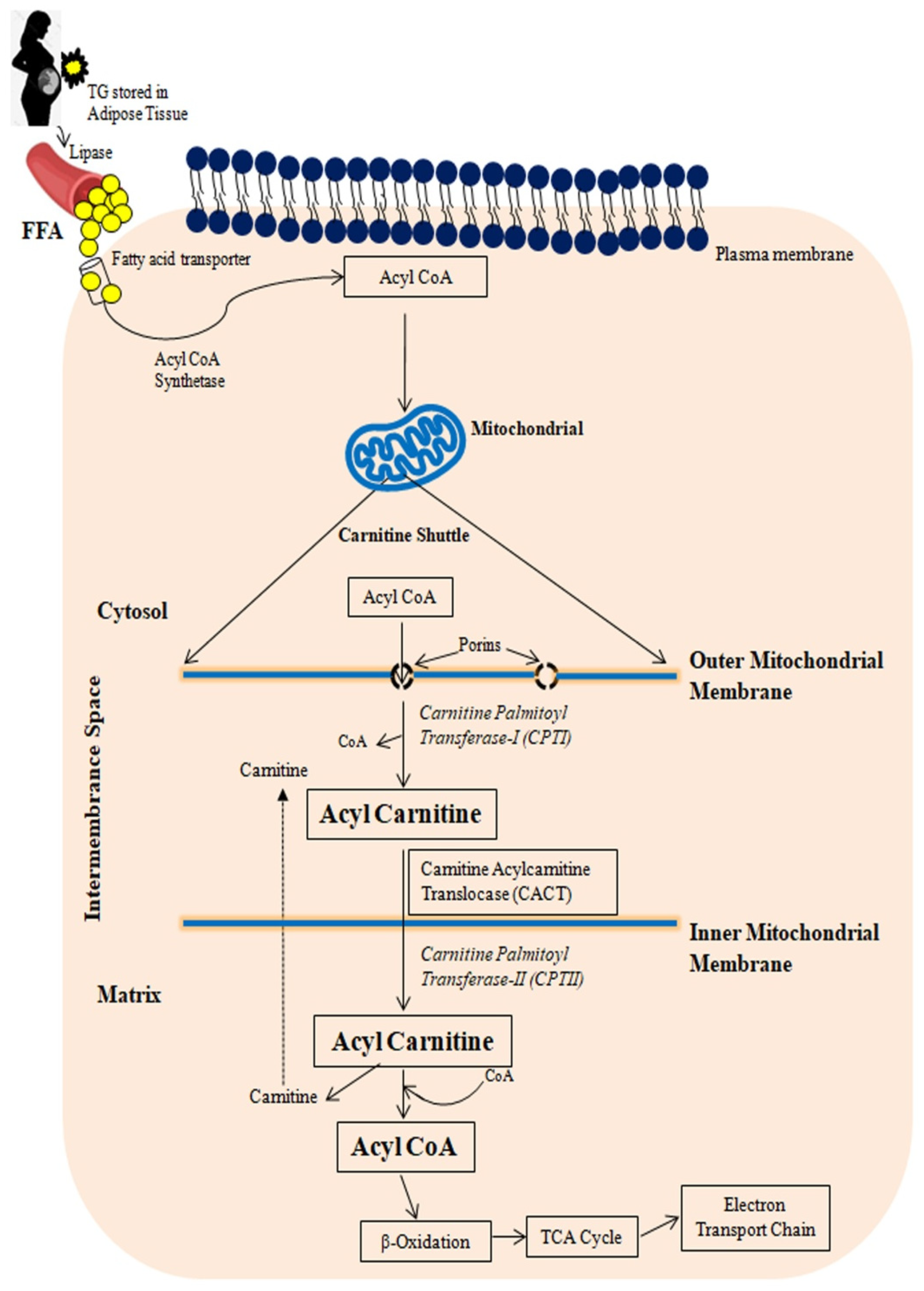
2.2.2. Mitochondrial β-Oxidation Cycle
Figure 2 depicts a schematic representation of mitochondrial fatty-acid oxidation and energy production. Mitochondrial β-oxidation is the primary oxidative mechanism for fatty acids including the oxidation of short-chain (<C6), medium-chain (C6–C12), and long-chain (>C12) fatty acids [45][26]. Long-chain fatty acids can only reach the mitochondria via CPT1 transport mechanism [46][27]. Short- and medium-chain fatty acids can freely enter the mitochondria. As a result, CPT1 in the mitochondria is the rate-limiting enzyme in β-oxidation.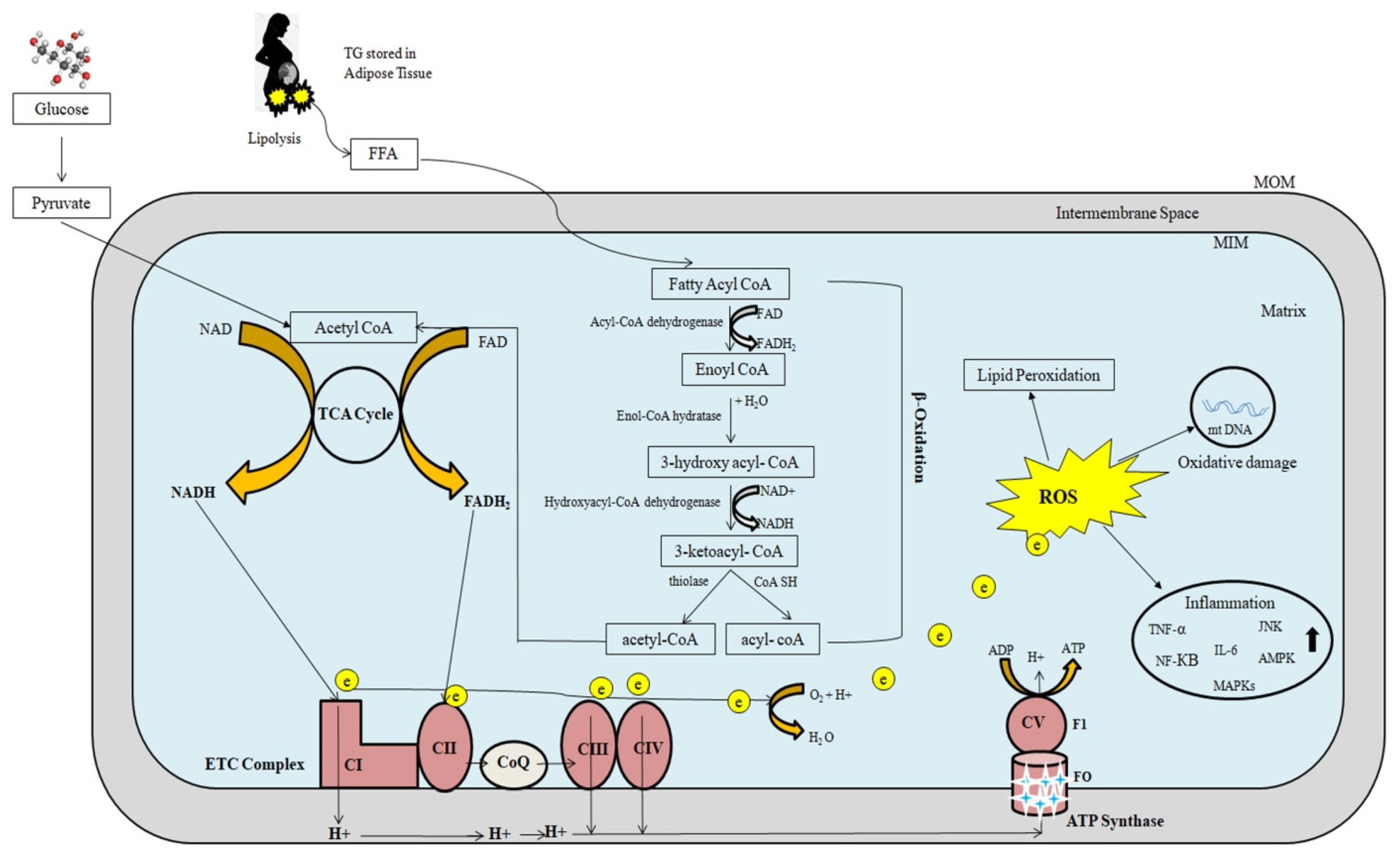
2.2.3. Oxidative Phosphorylation
The main source of cellular energy is the electron transport chain (ETC) and oxidative phosphorylation through critical activities of protein complexes in the inner mitochondrial membrane. High-energy electrons released during the citric acid cycle and β-oxidation are captured by nicotinamide adenine dinucleotide (NAD) and flavin adenine dinucleotide (FAD), resulting in NADH and FADH2, respectively [47][28]. NADH and FADH2 molecules donate these high-energy electrons to the ETC [48][29]. The transfer of electrons to O2 is an energy-yielding reaction by the passage of electrons through a series of carriers, which constitute the ETC. These carriers include four complexes (complex I, II, III, IV) in the inner mitochondrial membrane. A fifth protein complex (complex V), also in the inner mitochondrial membrane, then serves to couple the energy-yielding reactions of electron transport to ATP synthesis. Complex I receives electrons from NADH, while complex II receives electrons from FADH2. Complexes I and II provide electrons to Coenzyme Q (CoQ). CoQ (also called ubiquinone) is a small, lipid-soluble molecule that carries electrons through the MIM to complex III. Electrons are then transferred from complex III to cytochrome c, which then carries electrons to complex IV (cytochrome oxidase), where they are finally transferred to O2. Water is formed as a result of electron transfer from Complex IV to oxygen. At complexes I, III, and IV, free energy is released as electrons pass along the chain, which is utilized to pump protons from the mitochondrial matrix to the intermembranous region, forming a proton gradient. The potential energy stored in this gradient is then used by a fifth protein complex (complex V), which couples the flow of protons along the electrochemical gradient back across the MIM to the synthesis of ATP.2.3. Regulation of Mitochondrial Fatty-Acid Oxidation and Reactive Oxygen Species Formation
CPT1 is inhibited by malonyl-CoA, which is formed during the first step of the synthesis of FFAs from acetyl-CoA by acetyl-CoA carboxylase [32,35][21][24]. Insulin has been shown to increase malonyl-CoA synthesis, which inhibits CPTI. Glucagon, on the other hand, decreases malonyl-CoA synthesis, leading to an increase in β-oxidation [49][30]. FFAs are degraded into acetyl-CoA molecules, which can either be fully degraded to CO2 by the Krebs cycle or condensed into ketone bodies, which are re-oxidized in peripheral tissues during fasting [30,32][19][21]. Under normal circumstances, this process carefully controls energy storage and disposal; however, it is hampered in patients with fatty liver disease, causing oxidative stress [30,50,51][19][31][32]. Increased oxidative stress causes inflammation directly by activating a number of inflammatory-signaling pathways, including the NF-κB and JNK pathways, as well as indirectly by increasing the gene expression of inflammatory cytokines including TNF-α, TGF-β, and Fas ligand [52][33]. Reduced mitophagy leads to an accumulation of significantly damaged mitochondria, which causes cell necrosis and the release of mitochondrial damage-associated molecular patterns (DAMPs), which may promote liver inflammation [53][34]. ROS such as superoxide anions, peroxides, and others are generated in the cytosol by enzymes such as amino acid oxidases, cyclooxygenases, lipoxygenases, nitric oxide (NO) synthase, and xanthine oxidase [54,55,56][35][36][37]. By transferring a single electron from NADPH to molecular oxygen, it becomes NADPH oxidase, which is the key source of ROS in liver diseases and produces superoxide anions in the mitochondria [57,58,59][38][39][40]. Mitochondrial ROS activate AMPK [33,35][22][24] and mitogen-activated protein kinases (MAPKs), including c-Jun N-terminal kinase (JNK) [66][41]. AMPK induces PGC-1α and promotes glucose and fatty-acid oxidation. PGC-1α interacts with the peroxisome proliferator–activated receptor (PPAR) to increase mitochondrial fatty acid β-oxidation by inducing the expression of multiple fatty acid-metabolizing enzymes, such as CPT1 and acyl-CoA dehydrogenases [67][42]. By activating NRF2, H2O2 production by mitochondria activates AMPK, which regulates antioxidant enzyme expression [35][24]. Proinflammatory cytokines such as interleukin 6 (IL-6), tumor necrosis factor (TNF-α), and interleukin 1β (IL-1β) are also stimulated by ROS development. The presence of oxidative stress in cells may set off a chain reaction that contributes to increased mtDNA damage and increased mitochondrial dysfunction [68][43].2.4. Mitochondrial Fatty-Acid Oxidation Defects
The last three steps of long-chain fatty-acid oxidation are catabolized by MTP, a heterooctamer of 4 α- and 4 β- subunits associated with the inner mitochondrial membrane [69][44]. The long-chain 3-enoyl-CoA hydratase enzymatic activity resides in the α-subunit amino-terminal domain while the carboxy-terminal domain contains the LCHAD enzymatic activity. The long-chain 3-ketoacyl-CoA thiolase enzymatic activity resides in the β-subunit. Both MTP subunit genes, HADHA and HADHB, are localized to chromosome 2p23 [70][45], and share a bidirectional promoter [71][46]. MTP defects are recessively inherited and can manifest as either an isolated LCHAD deficiency or complete MTP deficiency, in which all three enzymes are deficient [72][47]. Infants born with these recessively inherited disorders typically present with nonketotic hypoglycemia and hepatic encephalopathy, which may progress to coma and death [73][48]. They can also present as unexpected death, cardiomyopathy, or slowly progressive myopathy and peripheral neuropathy [74,75][49][50]. A common mutation in exon 15 of the α-subunit, G1528C, which causes an amino acid change at position 474 in the LCHAD catalytic site from glutamic acid to glutamine (E474Q) [76,77][51][52].3. Mechanism of the Association between Fetal LCHAD Deficiency and AFLP
The precise mechanism for the association between fetal LCHAD-deficiency and maternal AFLP is not fully elucidated. Figure 3 depicts the likely mechanisms underlying the association between fetal LCHAD and maternal AFLP. Mitochondrial dysfunction and damage have been documented in children with LCHAD deficiency [89,90,91][53][54][55]. It is likely that hepatotoxic long-chain 3-hydroxyacyl fatty acid intermediates produced in the fetus due to blockages in the mitochondrial β-oxidation caused by the fetal LCHAD deficiency will accumulate in the maternal circulation causing liver injury and AFLP. It is also highly likely that the placenta is the major source for the 3-hydroxy fatty acid metabolites.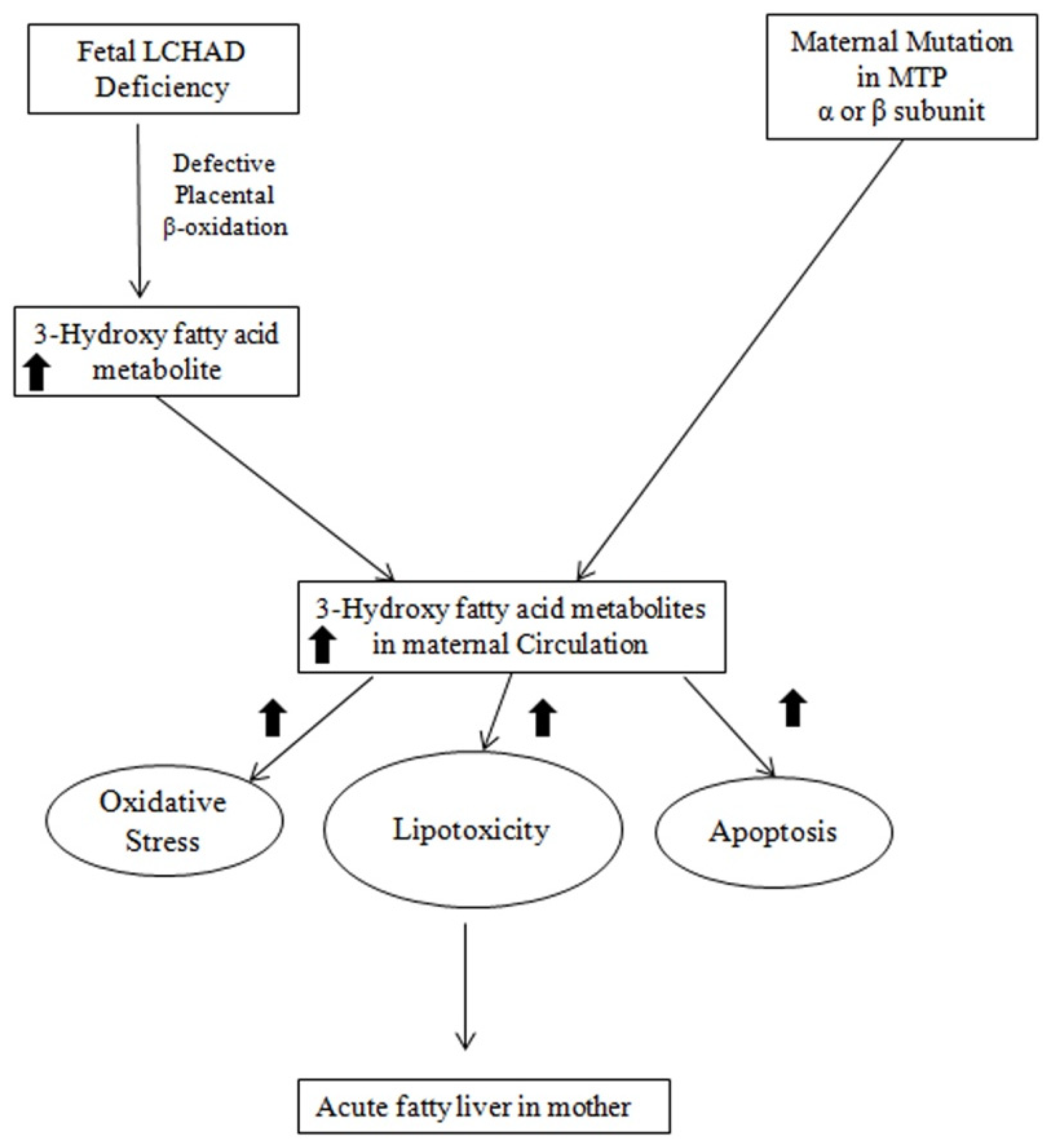
4. Conclusions
References
- Ibdah, J.A. Acute fatty liver of pregnancy: An update on pathogenesis and clinical implications. World J. Gastroenterol. 2006, 12, 7397–7404.
- Mjahed, K.; Charra, B.; Hamoudi, D.; Noun, M.; Barrou, L. Acute fatty liver of pregnancy. Arch. Gynecol. Obstet. 2006, 274, 349–353.
- Rolfes, D.B.; Ishak, K.G. Acute fatty liver of pregnancy: A clinicopathologic study of 35 cases. Hepatology 1985, 5, 1149–1158.
- Ko, H.; Yoshida, E.M. Acute fatty liver of pregnancy. Can. J. Gastroenterol. 2006, 20, 25–30.
- Zhou, H.; Shi, C.; Hu, S.; Zhu, H.; Ren, J.; Chen, Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis 2018, 21, 599–615.
- Jin, Q.; Li, R.; Hu, N.; Xin, T.; Zhu, P.; Hu, S.; Ma, S.; Zhu, H.; Ren, J.; Zhou, H. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018, 14, 576–587.
- Sumneang, N.; Siri-Angkul, N.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch. Biochem. Biophys. 2020, 680, 108241.
- Ding, M.; Ning, J.; Feng, N.; Li, Z.; Liu, Z.; Wang, Y.; Li, X.; Huo, C.; Jia, X.; Xu, R.; et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J. Pineal Res. 2018, 64, e12447.
- Feng, S.T.; Wang, Z.Z.; Yuan, Y.H.; Wang, X.L.; Sun, H.M.; Chen, N.H.; Zhang, Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res. 2020, 151, 104553.
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335.
- Kong, B.; Tsuyoshi, H.; Orisaka, M.; Shieh, D.B.; Yoshida, Y.; Tsang, B.K. Mitochondrial dynamics regulating chemoresistance in gynecological cancers. Ann. N. Y. Acad. Sci. 2015, 1350, 1–16.
- Ding, M.; Liu, C.; Shi, R.; Yu, M.; Zeng, K.; Kang, J.; Fu, F.; Mi, M. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol. 2020, 229, e13428.
- Jang, S.; Javadov, S. OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 2020, 51, 30–39.
- Cerveny, K.L.; Tamura, Y.; Zhang, Z.; Jensen, R.E.; Sesaki, H. Regulation of mitochondrial fusion and division. Trends Cell Biol. 2007, 17, 563–569.
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200.
- Zhang, L.; Han, L.; Ma, R.; Hou, X.; Yu, Y.; Sun, S.; Xu, Y.; Schedl, T.; Moley, K.H.; Wang, Q. Sirt3 prevents maternal obesity-associated oxidative stress and meiotic defects in mouse oocytes. Cell Cycle 2015, 14, 2959–2968.
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105.
- Du, K.; Ramachandran, A.; McGill, M.R.; Mansouri, A.; Asselah, T.; Farhood, A.; Woolbright, B.L.; Ding, W.X.; Jaeschke, H. Induction of mitochondrial biogenesis protects against acetaminophen hepatotoxicity. Food Chem. Toxicol. 2017, 108, 339–350.
- Pessayre, D.; Fromenty, B.; Berson, A.; Robin, M.A.; Lettéron, P.; Moreau, R.; Mansouri, A. Central role of mitochondria in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 34–87.
- Vacca, M.; Allison, M.; Griffin, J.L.; Vidal-Puig, A. Fatty Acid and Glucose Sensors in Hepatic Lipid Metabolism: Implications in NAFLD. Semin. Liver Dis. 2015, 35, 250–261.
- Gusdon, A.M.; Song, K.X.; Qu, S. Nonalcoholic Fatty liver disease: Pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027.
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135.
- Wang, L.; Scott, I.; Zhu, L.; Wu, K.; Han, K.; Chen, Y.; Gucek, M.; Sack, M.N. GCN5L1 modulates cross-talk between mitochondria and cell signaling to regulate FoxO1 stability and gluconeogenesis. Nat. Commun. 2017, 8, 523.
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320.
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S20–S27.
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199.
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007.
- Walsh, C.T.; Tu, B.P.; Tang, Y. Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism. Chem. Rev. 2018, 118, 1460–1494.
- Sun, F.; Zhou, Q.; Pang, X.; Xu, Y.; Rao, Z. Revealing various coupling of electron transfer and proton pumping in mitochondrial respiratory chain. Curr. Opin. Struct. Biol. 2013, 23, 526–538.
- Prentki, M.; Joly, E.; El-Assaad, W.; Roduit, R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: Role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes 2002, 51 (Suppl. S3), S405–S413.
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746.
- Larosche, I.; Lettéron, P.; Fromenty, B.; Vadrot, N.; Abbey-Toby, A.; Feldmann, G.; Pessayre, D.; Mansouri, A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J. Pharmacol. Exp. Ther. 2007, 321, 526–535.
- Feldstein, A.E. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin. Liver Dis. 2010, 30, 391–401.
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107.
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396.
- Song, X.; Li, T. Ripk3 mediates cardiomyocyte necrosis through targeting mitochondria and the JNK-Bnip3 pathway under hypoxia-reoxygenation injury. J. Recept. Signal Transduct. Res. 2019, 39, 331–340.
- Upadhyay, K.K.; Jadeja, R.N.; Vyas, H.S.; Pandya, B.; Joshi, A.; Vohra, A.; Thounaojam, M.C.; Martin, P.M.; Bartoli, M.; Devkar, R.V. Carbon monoxide releasing molecule-A1 improves nonalcoholic steatohepatitis via Nrf2 activation mediated improvement in oxidative stress and mitochondrial function. Redox Biol. 2020, 28, 101314.
- Xu, J.; Peng, Y.; Zeng, Y.; Hua, Y.Q.; Xu, X.L. 2, 3, 4’, 5-tetrahydroxystilbene-2-0-β-d Glycoside Attenuates Age- and Diet-Associated Non-Alcoholic Steatohepatitis and Atherosclerosis in LDL Receptor Knockout Mice and Its Possible Mechanisms. Int. J. Mol. Sci. 2019, 20, 1617.
- Knebel, B.; Fahlbusch, P.; Dille, M.; Wahlers, N.; Hartwig, S.; Jacob, S.; Kettel, U.; Schiller, M.; Herebian, D.; Koellmer, C.; et al. Fatty Liver Due to Increased. Front. Cell Dev. Biol. 2019, 7, 248.
- Erland, L.A.E.; Shukla, M.R.; Singh, A.S.; Murch, S.J.; Saxena, P.K. Melatonin and serotonin: Mediators in the symphony of plant morphogenesis. J. Pineal Res. 2018, 64, e12452.
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024.
- Fromenty, B.; Pessayre, D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154.
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535.
- Kamijo, T.; Wanders, R.J.; Saudubray, J.M.; Aoyama, T.; Komiyama, A.; Hashimoto, T. Mitochondrial trifunctional protein deficiency. Catalytic heterogeneity of the mutant enzyme in two patients. J. Clin. Investig. 1994, 93, 1740–1747.
- Aoyama, T.; Wakui, K.; Orii, K.E.; Hashimoto, T.; Fukushima, Y. Fluorescence in situ hybridization mapping of the alpha and beta subunits (HADHA and HADHB) of human mitochondrial fatty acid beta-oxidation multienzyme complex to 2p23 and their evolution. Cytogenet. Genome Res. 1997, 79, 221–224.
- Orii, K.E.; Orii, K.O.; Souri, M.; Orii, T.; Kondo, N.; Hashimoto, T.; Aoyama, T. Genes for the human mitochondrial trifunctional protein alpha- and beta-subunits are divergently transcribed from a common promoter region. J. Biol. Chem. 1999, 274, 8077–8084.
- Ibdah, J.A.; Bennett, M.J.; Rinaldo, P.; Zhao, Y.; Gibson, B.; Sims, H.F.; Strauss, A.W. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N. Engl. J. Med. 1999, 340, 1723–1731.
- Rinaldo, P.; Raymond, K.; al-Odaib, A.; Bennett, M.J. Clinical and biochemical features of fatty acid oxidation disorders. Curr. Opin. Pediatr. 1998, 10, 615–621.
- Pons, R.; Roig, M.; Riudor, E.; Ribes, A.; Briones, P.; Ortigosa, L.; Baldellou, A.; Gil-Gibernau, J.; Olesti, M.; Navarro, C.; et al. The clinical spectrum of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr. Neurol. 1996, 14, 236–243.
- Ibdah, J.A.; Tein, I.; Dionisi-Vici, C.; Bennett, M.J.; IJlst, L.; Gibson, B.; Wanders, R.J.; Strauss, A.W. Mild trifunctional protein deficiency is associated with progressive neuropathy and myopathy and suggests a novel genotype-phenotype correlation. J. Clin. Investig. 1998, 102, 1193–1199.
- Sims, H.F.; Brackett, J.C.; Powell, C.K.; Treem, W.R.; Hale, D.E.; Bennett, M.J.; Gibson, B.; Shapiro, S.; Strauss, A.W. The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc. Natl. Acad. Sci. USA 1995, 92, 841–845.
- IJlst, L.; Wanders, R.J.; Ushikubo, S.; Kamijo, T.; Hashimoto, T. Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim. Biophys. Acta 1994, 1215, 347–350.
- Matern, D.; Schehata, B.M.; Shekhawa, P.; Strauss, A.W.; Bennett, M.J.; Rinaldo, P. Placental floor infarction complicating the pregnancy of a fetus with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Mol. Genet. Metab. 2001, 72, 265–268.
- Cecatto, C.; Godoy, K.D.S.; da Silva, J.C.; Amaral, A.U.; Wajner, M. Disturbance of mitochondrial functions provoked by the major long-chain 3-hydroxylated fatty acids accumulating in MTP and LCHAD deficiencies in skeletal muscle. Toxicol. In Vitro 2016, 36, 1–9.
- Steinmann, D.; Knab, J.; Priebe, H.J. Perioperative management of a child with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Paediatr. Anaesth. 2010, 20, 371–373.
- Tonin, A.M.; Grings, M.; Busanello, E.N.; Moura, A.P.; Ferreira, G.C.; Viegas, C.M.; Fernandes, C.G.; Schuck, P.F.; Wajner, M. Long-chain 3-hydroxy fatty acids accumulating in LCHAD and MTP deficiencies induce oxidative stress in rat brain. Neurochem. Int. 2010, 56, 930–936.
- Neuman-Łaniec, M.; Wierzba, J.; Irga, N.; Zaborowska-Sołtys, M.; Balcerska, A. LCHAD (long-chain 3-hydroxyacyl-CoA dehydrogenase) deficiency as a cause of sudden death of a three months old infant. Med. Wieku Rozw. 2002, 6, 221–226.
- Olpin, S.E.; Clark, S.; Andresen, B.S.; Bischoff, C.; Olsen, R.K.; Gregersen, N.; Chakrapani, A.; Downing, M.; Manning, N.J.; Sharrard, M.; et al. Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J. Inherit. Metab. Dis. 2005, 28, 533–544.
- Natarajan, S.K.; Thangaraj, K.R.; Eapen, C.E.; Ramachandran, A.; Mukhopadhya, A.; Mathai, M.; Seshadri, L.; Peedikayil, A.; Ramakrishna, B.; Balasubramanian, K.A. Liver injury in acute fatty liver of pregnancy: Possible link to placental mitochondrial dysfunction and oxidative stress. Hepatology 2010, 51, 191–200.
- Natarajan, S.K.; Thomas, S.; Ramamoorthy, P.; Basivireddy, J.; Pulimood, A.B.; Ramachandran, A.; Balasubramanian, K.A. Oxidative stress in the development of liver cirrhosis: A comparison of two different experimental models. J. Gastroenterol. Hepatol. 2006, 21, 947–957.