According to the Health Physics Society, radiation is defined as energy that travels from a source through space as waves or particles and it can penetrate different materials. The electromagnetic spectrum consists of different wavelengths and frequencies, in which non-ionizing (low frequency) and ionizing (high frequency) radiation are found. Gene expression will be changed due to radiation.
- breast cancer model
- radiation
- gene expression
- estrogens
1. Introduction
Two types of radiation can be distinguished: non-ionizing and ionizing radiation; the first radiation comprises optical radiation and electromagnetic fields [1]. The optical category is divided into ultraviolet, visible, and infra-red subcategories, while the electromagnetic one can be subdivided depending on the radiofrequency [1][2]. Non-ionizing radiation can be obtained from several natural sources like the Sun and lighting, or man-made sources as those used in industrial/medical applications and wireless communications [1]. On average, a person receives approximately 2.4 mGy of natural-originated radiation every year, and this can vary according to their geographical location, for instance, countries such as Brazil, India, and China are among those with high levels of terrestrial radiation [3][4]. Ionizing radiation comprises electrically charged particles (ions), positive ones such as alpha particles and negative ones such as electrons [2]. This radiation was found after the discovery of X-rays in 1895 [5].
According to the Advisory Committee on Human Radiation Experiments alpha, beta, and gamma/X-ray radiation are the most known ionizing radiation; alpha particles are formed by two neutrons and two protons from the nucleus of the atom during the decrease of the atomic mass number and reduction of the atomic number; it results from the radioactive decay of heavy elements such as plutonium, radium, or thorium and its weight does not allow them to travel far away, being stopped by a piece of paper, and although these particles cannot pass through paper or our skin, if they are released into the body from a radioactive source, they can affect cells in our body, damaging the cells and the DNA [6]. Unlike alpha particles, beta particles are negatively charged when emitted during radioactive decay [7]. Even though these particles can reach longer distances, they can be blocked by a thin layer of substance; however, if they are swallowed or inhaled the damage can be high as that caused by alpha particles [7]. Regarding gamma rays, this radiation is frequently emitted during the radioactive decay along with alpha or beta particles, they are high-energy photons. Gamma radiation is high-energy electromagnetic radiation emitted along with alpha and beta particles during radioactive decay. Gamma particles are pure energy. Different from alpha and beta particles, gamma particles can easily penetrate the skin and cause serious tissue and DNA damage [7]. Similarly, X-rays are also pure energy but are emitted from parts of the atom different from the nucleus. This radiation is widely used in the medical field and industrial processes [6][7]. Other types of ionizing radiation can be found, such as cosmic radiation that penetrates our atmosphere and comprises mainly protons, alpha particles, and heavier atomic nuclei [8].
2. Gene Expression Induced by Radiation
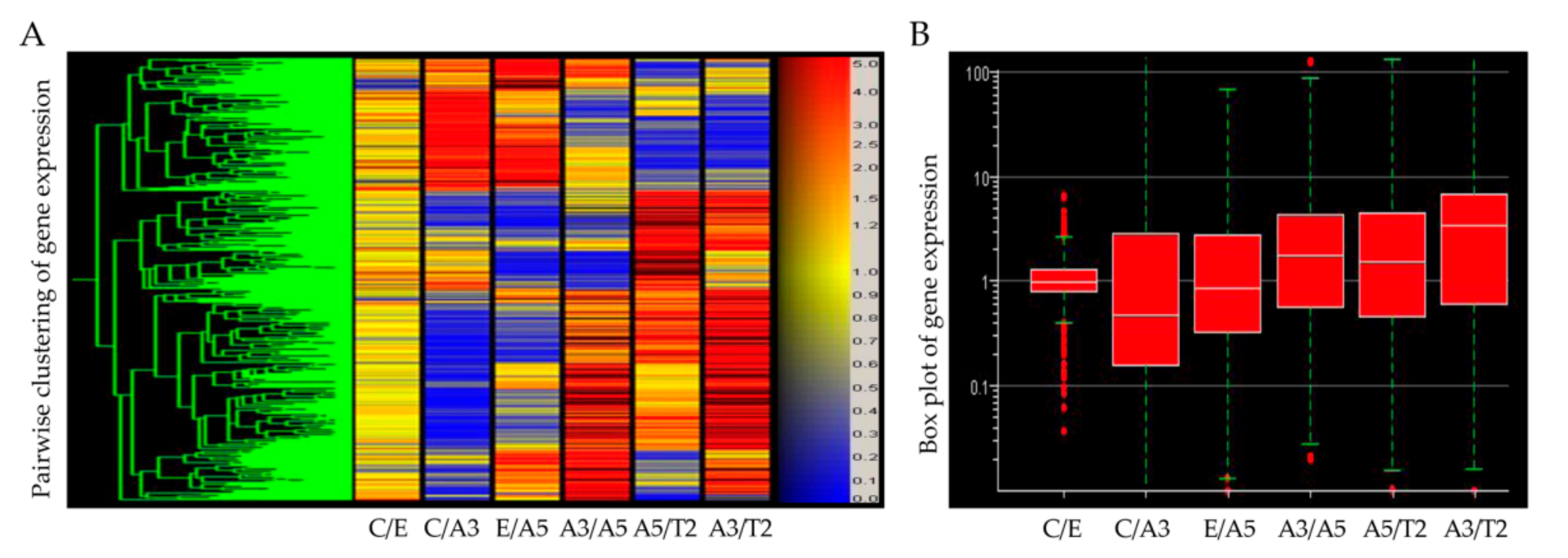
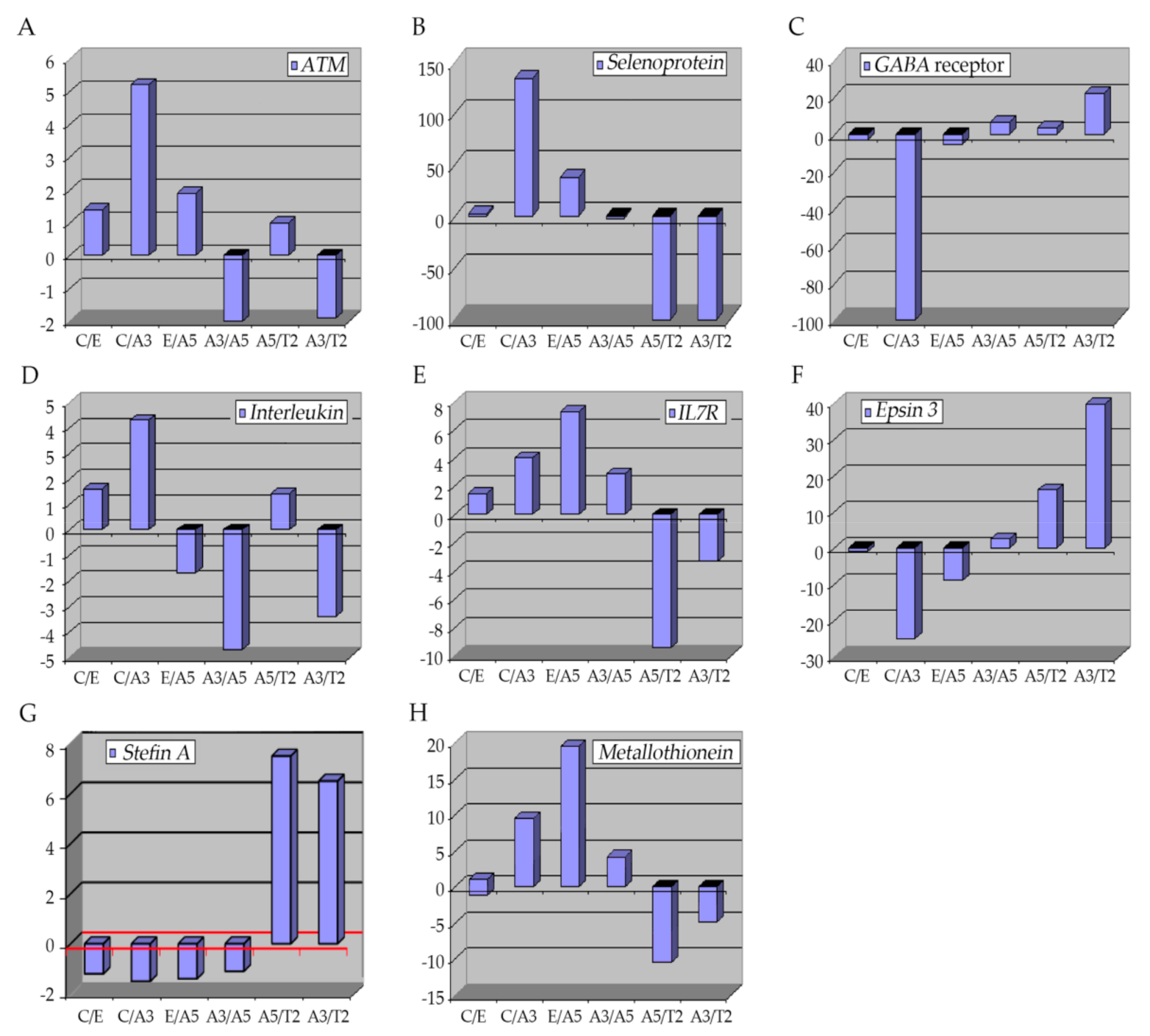
2.1. The Ataxia-Telangiectasia Mutated Gene
The ataxia-telangiectasia mutated gene (ATM) encodes for a 350 kDa protein serine/threonine kinase, key in the DNA-damage response elements since it can detect double-strand breaks (DSBs), fundamental in the cell-cycle checkpoint controlling [17][18][19][20]. The ATM gene has 66 exons with approximately 150 kb of genomic expansion [18]. Mutations of the ATM gene can explain ataxia telangiectasia (AT), a rare neurodegenerative disease, which is manifested clinically by skin and ocular telangiectasia, immunological deficiency, neuronal deficiency, sino-pulmonary infections, cellular sensitivity to ionizing radiation, and predisposition to cancer [21][22][23][24][25][26][27]. However, it has been estimated that approximately 2% of the adult population presents the heterozygosity for an ATM variant [28][29][30]. Even though this group exhibits no phenotypical abnormalities, it has been reported that ATM heterozygotes have a high risk of developing breast cancer, with about a 5-fold increase compared with the general population [23][31][32][33][34][35][36]. Normally, ATM expression is down-regulated in breast cancer tissues [36]. Locally advanced breast tumors have shown a reduced expression of ATM by epigenetic silencing [37].2.2. Selenoproteins (SEPP1)
These are metalloproteins with certain characteristics that allow them to have a high affinity to metal; they present a specific amino acid, seleno-cysteine (Se-Cys) [38]. There are 25 types of selenoproteins in humans with this characteristic [39][40]. Selenoproteins are involved in several cellular processes related to metastasis, comprising cell adhesion, matrix degradation, migration, invasion, and proliferation [41]. Among these processes, roles in redox balance and calcium equilibrium have been described [41], but the metabolism of these proteins can also modify signaling pathways in cancer cells [42]. Glutathione peroxidases (GPXs), are selenoproteins that are responsible for the protection of tissues against reductions derived from the action of ROS such as those produced during ionizing radiation [43]. These enzymes are in charge of the hydroperoxide (H2O2) reduction, a type of ROS produced in the cell [44][45], thus decreasing the negative effects of ROS and contributing to the anti-metastatic function [41][46][47][48]. They are also involved in roles such as DNA-repairing and cytokine control, thus, supplements with Se have been used in chemotherapy since it was reported that it increased the selenoprotein expression in plasma [49]. Similarly, other selenoproteins that participate in redox processes during tumor progression are the thioredoxin reductases (TXNRDs) with three subtypes placed in the cytosol and nucleus, mitochondria, and sperm [50]. This selenoprotein subfamily is up-regulated in several cancers such as lung cancer [51][52], breast cancer [53][54], and astrocytomas [55].2.3. GABA Receptor
γ-Aminobutyric acid (GABA) is the principal inhibitor in the central nervous system and it is present in the peripheral nervous system as well [56][57]. The expression of GABA and GABA receptors (GABARAP) is mainly found in brain structures; however, its expression can be detected in other organs like the pancreas, kidney, intestine, prostate, testis, ovaries, and liver where it can trigger hormone and neuronal activity [56]. Ionizing radiation augments the GABA receptors mRNA in C17.2 mouse neural stem-like cell lines and in mouse primary neural stem cells, presumably by altering the neuronal function [58]. Nevertheless, other studies have shown contrary effects that may be because of the differences in doses and time of exposure [58][59][60]. It was reported that the GABA signaling pathway was altered in some cancers such as pancreatic, gastric, and breast cancers, an increment in the expression of GABA and GABA receptors was found [61][62][63]. It was observed that activation of these receptors could decrease cell proliferation and migration [64][65][66]. Thus, the GABARAP system was suggested to play a role in tumorigenesis as a cell migration and proliferation inhibitor [67].2.4. Interleukins
Interleukins (IL) are proteins secreted primarily by CD3+ and CD4+. They belong to the cytokine superfamily with about 38 different types of IL and are responsible for the interactions between cells [68]. In tumors, cytokines collaborate with different elements such as cancer stem cells, microRNA, epithelial-mesenchymal transition (EMT) markers, and transcription factors, thus these biomolecules are involved in different processes, principally in systemic inflammation and immune system modulation, involving cell migration, proliferation, maturation, and adhesion necessary for the inflammatory response [69][70].2.5. Epsins 3
Epsins are adaptor proteins, part of a family of ubiquitin-binding endocytic proteins [71]. In mammals, three genes encode for each isoform; epsin1, epsin2, and epsin3 [72][73][74]. These proteins have a well conserved NH2-terminal homology domain (ENTH), important in ubiquitination and the ENTH portion comprises about 150 amino acids and it is essential for binding inositol phospholipids and proteins [75]. Thus, involved in signaling pathways like Notch, Rho GTPase, and VEGFRs [76][77][78][79]. Epsin type 3 is predominately expressed in the stomach and the epithelia while types 1 and 2 are expressed in different cell types, with no specific location and repetitious functions [72][73][74]. Besides, it has been shown that these proteins are up-regulated in different cancers [80][81]. In breast cancer, Epsin proteins induce NF-kB, which is fundamental in developing the disease, high levels of epsins are associated with low relapse-free survival rates, especially in ER-negative breast cancer types [71]. Epsin3 acts as an oncogene in ER-positive breast and other cancers such as non-small cell lung cancer [82]. Similarly, the epsin3 protein has been identified in glioblastoma cell lines and samples of patients with glioma, and its overexpression has been shown to induce cell migration and invasion through transcription factors such as Slug, Twist, and ZEB1 promoting EMT in these glioma-type cells [82]. Studies in the breast cancer model indicated that A3, characterized by cells transformed by radiation only, had higher epsin gene expression than C.2.6. Stefin A (Cystatin A)
To understand the role of stefin genes (cystatin A, CSTA), it is important to know that cathepsins are lysosomal proteases and they can be classified into serine, cysteine, and aspartyl cathepsins [83][84], with 11 well-identified types: B, H, L, S, C, K, O, F, V, W, and X/Z [85]. These proteases are essential in protein degradation processes, associated with phagocytosis, endocytosis, and autophagy [86], in addition to apoptosis, immune response, development, differentiation, and pro-tumorigenic functions [83][85][87]. For instance, cathepsin S type is involved in tumor progression [88], angiogenesis, tumor growth [89][90], similarly, cathepsin L is involved in neovascularization [91], migration, and invasion processes as well [92][93]. Alteration and changes in expression of cathepsins are associated with pathological circumstances, for example, they are secreted into the extracellular medium in cancer [94][95][96].2.7. Metallothioneins
Metallothioneins (MTs) are small (approximately 6–7 kDa) cytosolic proteins with a high content of cysteine groups (30%) [97][98]. There are four main MT isoforms in humans—MT1, MT2, MT3, and MT4—encoded by a gene located at the 16q13 locus [99]. There is evidence that connects MTs with tumor formation, progression, and drug resistance [99]. Their principal role is in homeostasis and the detoxification of heavy metals, oxidative stress, and DNA damage processes [99][100]. They bind to heavy metals (with great affinity) via the thiol binding part of the cysteine-enriched portion [99]. When MTs bind to metals such as zinc and copper, they can regulate different important processes such as cell growth, proliferation, differentiation, metastasis, and protection against oxidative radicals produced by drugs and radiation [99][101][102][103]. However, they also can bind to other metals like cadmium, mercury, and platinum, among others, to protect cells from these heavy metals [104]. The expression of MTs depends on the type of tumor suggesting a specific role in carcinogenesis [97][105][106][107]. MTs have been reported to be up-regulated in breast, ovarian urinary bladder, and nasopharyngeal cancer, and melanoma [108][109][110][111][112]. A positive correlation has been established between MTs and Ki-67, a marker of cellular proliferation in breast cancer [108][113][114]. Specific MTs such as MT1F and MT2A have been found in greater numbers in cancer in stage 3 than in stage 1 and 2 when histological samples of breast cancer were analyzed [114][115]. In addition, zinc was demonstrated to increase the expression of the vascular epithelial growth factor (VEGF) in three breast cell lines [116].3. Conclusions
References
- Ng, K.H. Non-Ionizing radiations—sources, biological effects, emissions and exposures. Electromagnetic Field sand Our Health. In Proceedings of the International Conferenceon Non-Ionizing Radiation at UNITEN(ICNIR2003), Kuala Lumpur, Malaysia, 20–22 October 2003.
- Baba, A.I.; Catoi, C. Carcinogenesis. In Comparative Oncology; The Publishing House of the Romanian: Bucharest, Romania, 2007.
- United Nations Scientific Committee on the Effects of Atomic Radiation. Sources and Effects of Ionizing Radiation: UNSCEAR 2008 Report to the General Assembly Scientific Annexes A and B; United Nations: New York, NY, USA, 2010; p. 683.
- Hendry, J.H.; Simon, S.L.; Wojcik, A.; Sohrabi, M.; Burkart, W.; Cardis, E.; Laurier, D.; Tirmarche, M.; Hayata, I. Human exposure to high natural background radiation: What can it teach us about radiation risks? J. Radiol. Prot. 2009, 29, A29–A42.
- Parkin, D.M.; Darby, S.C. 12. Cancers in 2010 attributable to ionising radiation exposure in the UK. Br. J. Cancer 2011, 105, S57–S65.
- Keith, S.; Doyle, J.R.; Harper, C.; Mumtaz, M.; Tarrago, O.; Wohlers, D.W.; Diamond, G.L.; Citra, M.; Barber, L.E. Toxicological Profile for Radon; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2012; p. 283.
- EPA. Radiation Basics. United States Environmental Protection Agency. Available online: https://www.epa.gov/radiation/radiation-basics (accessed on 10 May 2021).
- USNRC. Radiation Basics. United States Nuclear Regulatory Commission. Available online: https://www.nrc.gov/about-nrc/radiation/health-effects/radiation-basics.html (accessed on 10 May 2021).
- Sharungbam, G.D.; Schwager, C.; Chiblak, S.; Brons, S.; Hlatky, L.; Haberer, T.; Debus, J.; Abdollahi, A. Identification of stable endogenous control genes for transcriptional profiling of photon, proton and carbon-ion irradiated cells. Radiat. Oncol. 2012, 7, 70.
- Little, J.B.; Loeb, K.R.; Loeb, L.A. Radiation carcinogenesis. Carcinogenesis 2000, 21, 397–404.
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750.
- Zhou, Y.-C.; Liu, J.-Y.; Li, J.; Zhang, J.; Xu, Y.-Q.; Zhang, H.-W.; Qiu, L.-B.; Ding, G.-R.; Su, X.-M.; Shi, M.; et al. Ionizing Radiation Promotes Migration and Invasion of Cancer Cells Through Transforming Growth Factor-Beta–Mediated Epithelial–Mesenchymal Transition. Int. J. Radiat. Oncol. 2011, 81, 1530–1537.
- Fujita, M.; Otsuka, Y.; Yamada, S.; Iwakawa, M.; Imai, T. X-ray irradiation and Rho-kinase inhibitor additively induce invasiveness of the cells of the pancreatic cancer line, MIAPaCa-2, which exhibits mesenchymal and amoeboid motility. Cancer Sci. 2011, 102, 792–798.
- Sofia Vala, I.; Martins, L.R.; Imaizumi, N.; Nunes, R.J.; Rino, J.; Kuonen, F.; Carvalho, L.M.; Ruegg, C.; Grillo, I.M.; Barata, J.T.; et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS ONE 2010, 5, e11222.
- Pickhard, A.C.; Margraf, J.; Knopf, A.; Stark, T.; Piontek, G.; Beck, C.; Boulesteix, A.-L.; Scherer, E.Q.; Pigorsch, S.; Schlegel, J.; et al. Inhibition of radiation induced migration of human head and neck squamous cell carcinoma cells by blocking of EGF receptor pathways. BMC Cancer 2011, 11, 388.
- Calaf, G.M.; Roy, D.; Narayan, G.; Balajee, A.S. Differential expression of cell adhesion molecules in an ionizing radiation-induced breast cancer model system. Oncol. Rep. 2013, 30, 285–291.
- Angèle, S.; Treilleux, I.; Tanière, P.; Martel-Planche, G.; Vuillaume, M.; Bailly, C.; Brémond, A.; Montesano, R.; Hall, J. Abnormal expression of the ATM and TP53 genes in sporadic breast carcinomas. Clin. Cancer Res. 2000, 6, 3536–3544.
- Prokopcova, J.; Kleibl, Z.; Banwell, C.M.; Pohlreich, P. The role of ATM in breast cancer development. Breast Cancer Res. Treat. 2006, 104, 121–128.
- Cuatrecasas, M.; Santamaria, G.; Velasco, M.; Camacho, E.; Hernandez, L.; Sanchez, M.; Orrit, C.; Murcia, C.; Cardesa, A.; Campo, E.; et al. ATM gene expression is associated with differentiation and angiogenesis in infiltrating breast carcinomas. Histol. Histopathol. 2005, 21, 149–156.
- Kitagawa, R.; Kastan, M. The ATM-dependent DNA Damage Signaling Pathway. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 99–109.
- Teive, H.A.; Moro, A.; Moscovich, M.; Arruda, W.O.; Munhoz, R.P.; Raskin, S.; Ashizawa, T. Ataxia-telangiectasia—A historical review and a proposal for a new designation: ATM syndrome. J. Neurol. Sci. 2015, 355, 3–6.
- Thompson, D.; Duedal, S.; Kirner, J.; McGuffog, L.; Last, J.; Reiman, A.; Byrd, P.; Taylor, M.; Easton, D.F. Cancer Risks and Mortality in Heterozygous ATM Mutation Carriers. J. Natl. Cancer Inst. 2005, 97, 813–822.
- Furtado, S.; Das, S.; Suchowersky, O. A review of the inherited ataxias: Recent advances in genetic, clinical and neuropathologic aspects. Park. Relat. Disord. 1998, 4, 161–169.
- Morrell, D.; Cromartie, E.; Swift, M. Mortality and Cancer Incidence in 263 Patients With Ataxia-Telangiectasia2. J. Natl. Cancer Inst. 1986, 77, 89–92.
- Levy, A.; Lang, A.E. Ataxia-telangiectasia: A review of movement disorders, clinical features, and genotype correlations. Mov. Disord. 2018, 33, 1238–1247.
- Su, Y.; Swift, M. Mortality rates among carriers of ataxia-telangiectasia mutant alleles. Ann. Intern. Med. 2000, 133, 770–778.
- Dombernowsky, S.L.; Weischer, M.; Allin, K.H.; Bojesen, S.E.; Tybjjrg-Hansen, A.; Nordestgaard, B.G. Risk of Cancer by ATM Missense Mutations in the General Population. J. Clin. Oncol. 2008, 26, 3057–3062.
- Swift, M.; Reitnauer, P.J.; Morrell, D.; Chase, C.L. Breast and Other Cancers in Families with Ataxia-Telangiectasia. N. Engl. J. Med. 1987, 316, 1289–1294.
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am. J. Hum. Genet. 1986, 39, 573–583.
- Swift, M.; Morrell, D.; Massey, R.B.; Chase, C.L. Incidence of Cancer in 161 Families Affected by Ataxia–Telangiectasia. N. Engl. J. Med. 1991, 325, 1831–1836.
- Chessa, L.; Lisa, A.; Fiorani, O.; Zei, G. Ataxia-telangiectasia in Italy: Genetic analysis. Int. J. Radiat. Biol. 1994, 66, S31–S33.
- Renwick, A.; The Breast Cancer Susceptibility Collaboration (UK); Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875.
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257.
- Van Os, N.; Roeleveld, N.; Weemaes, C.; Jongmans, M.; Janssens, G.O.R.J.; Taylor, A.; Hoogerbrugge, N.; Willemsen, M. Health risks for ataxia-telangiectasia mutated heterozygotes: A systematic review, meta-analysis and evidence-based guideline. Clin. Genet. 2016, 90, 105–117.
- Marabelli, M.; Cheng, S.-C.; Parmigiani, G. Penetrance ofATMGene Mutations in Breast Cancer: A Meta-Analysis of Different Measures of Risk. Genet. Epidemiol. 2016, 40, 425–431.
- Foroughizadeh, M.; Mozdarani, H.; Majidzadeh-A, K.; Kaviani, A. Variation of ATM Gene Expression in Peripheral Blood Cells of Sporadic Breast Carcinomas in Iranian Patients. Avicenna J. Med. Biotechnol. 2012, 4, 95–101.
- Vo, Q.N.; Kim, W.-J.; Cvitanovic, L.; Boudreau, D.A.; Ginzinger, D.G.; Brown, K.D. The ATM gene is a target for epigenetic silencing in locally advanced breast cancer. Oncogene 2004, 23, 9432–9437.
- Handa, E.; Puspitasari, I.M.; Abdulah, R.; Yamazaki, C.; Kameo, S.; Nakano, T.; Koyama, H. Recent advances in clinical studies of selenium supplementation in radiotherapy. J. Trace Elem. Med. Biol. 2020, 62, 126653.
- Kryukov, G.; Castellano, S.; Novoselov, S.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of Mammalian Selenoproteomes. Science 2003, 300, 1439–1443.
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241.
- Marciel, M.P.; Hoffmann, P.R. Selenoproteins and Metastasis. Adv. Cancer Res. 2017, 136, 85–108.
- Gopalakrishna, R.; Gundimeda, U. Protein Kinase C as a Molecular Target for Cancer Prevention by Selenocompounds. Nutr. Cancer 2001, 40, 55–63.
- Hosseinimehr, S.J. The protective effects of trace elements against side effects induced by ionizing radiation. Radiat. Oncol. J. 2015, 33, 66–74.
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303.
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254.
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Peter, S. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896.
- Wu, W.-S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705.
- Muecke, R.; Schomburg, L.; Buentzel, J.; Kisters, K.; Micke, O.; German Working Group Trace Elements and Electrolytes in Oncology-AKTE. Selenium or No Selenium- That Is the Question in Tumor Patients: A New Controversy. Integr. Cancer Ther. 2010, 9, 136–141.
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346 Pt 1, 1–8.
- Fernandes, A.P.; Capitanio, A.; Selenius, M.; Brodin, O.; Rundlöf, A.-K.; Björnstedt, M. Expression profiles of thioredoxin family proteins in human lung cancer tissue: Correlation with proliferation and differentiation. Histopathology 2009, 55, 313–320.
- Soini, Y.; Kahlos, K.; Näpänkangas, U.; Kaarteenaho-Wiik, R.; Säily, M.; Koistinen, P.; Pääakkö, P.; Holmgren, A.; Kinnula, V.L. Widespread expression of thioredoxin and thioredoxin reductase in non-small cell lung carcinoma. Clin. Cancer Res. 2001, 7, 1750–1757.
- Cañas, A.; López-Sánchez, L.M.; Valverde-Estepa, A.; Hernández, V.; Fuentes, E.; Muñoz-Castañeda, J.R.; López-Pedrera, C.; Rodríguez, J.R.D.L.H.; Aranda, E.; Rodríguez-Ariza, A. Maintenance of S-nitrosothiol homeostasis plays an important role in growth suppression of estrogen receptor-positive breast tumors. Breast Cancer Res. 2012, 14, R153.
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010, 12, R44.
- Esen, H.; Erdi, F.; Kaya, B.; Feyzioglu, B.; Keskin, F.; Demir, L.S.; Feyzioglu, B. Tissue thioredoxin reductase-1 expression in astrocytomas of different grades. J. Neuro-Oncol. 2014, 121, 451–458.
- Brzozowska, A.; Burdan, F.; Duma, D.; Solski, J.; Mazurkiewicz, M. γ-amino butyric acid (GABA) level as an overall survival risk factor in breast cancer. Ann. Agric. Environ. Med. 2017, 24, 435–439.
- Serrano-Regal, M.P.; Bayón-Cordero, L.; Ordaz, R.P.; Garay, E.; Limon, A.; Arellano, R.O.; Matute, C.; Sánchez-Gómez, M.V. Expression and Function of GABA Receptors in Myelinating Cells. Front. Cell. Neurosci. 2020, 14, 256.
- Eom, H.S.; Park, H.R.; Jo, S.K.; Kim, Y.S.; Moon, C.; Kim, S.-H.; Jung, U. Ionizing Radiation Induces Altered Neuronal Differentiation by mGluR1 through PI3K-STAT3 Signaling in C17.2 Mouse Neural Stem-Like Cells. PLoS ONE 2016, 11, e0147538.
- Wu, P.H.; Coultrap, S.; Pinnix, C.; Davies, K.D.; Tailor, R.; Ang, K.K.; Browning, M.D.; Grosshans, D.R. Radiation Induces Acute Alterations in Neuronal Function. PLoS ONE 2012, 7, e37677.
- Lang, M.; Moradi-Chameh, H.; Zahid, T.; Gane, J.; Wu, C.; Valiante, T.A.; Zhang, L. Regulating hippocampal hyperexcitability through GABAB Receptors. Physiol. Rep. 2014, 2, e00278.
- Takehara, A.; Hosokawa, M.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Nakamura, Y.; Nakagawa, H. γ-Aminobutyric Acid (GABA) Stimulates Pancreatic Cancer Growth through Overexpressing GABAA Receptor π Subunit. Cancer Res. 2007, 67, 9704–9712.
- Matuszek, M.; Jesipowicz, M.; Kleinrok, Z. GABA content and GAD activity in gastric cancer. Med. Sci. Monit. 2001, 7, 377–381.
- Papadopoulos, V.; Kapsis, A.; Li, H.; Amri, H.; Hardwick, M.; Culty, M.; Kasprzyk, P.G.; Carlson, M.; Moreau, J.P.; Drieu, K. Drug-induced inhibition of the peripheral-type benzodiazepine receptor expression and cell proliferation in human breast cancer cells. Anticancer Res. 2000, 20, 2835–2847.
- Schuller, H.M.; Al-Wadei, H.A.; Majidi, M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis 2008, 29, 1979–1985.
- Wang, T.; Huang, W.; Chen, F. Baclofen, a GABAB receptor agonist, inhibits human hepatocellular carcinoma cell growth in vitro and in vivo. Life Sci. 2008, 82, 536–541.
- Opolski, A.; Mazurkiewicz, M.; Wietrzyk, J.; Kleinrok, Z.; Radzikowski, C. The role of GABA-ergic system in human mammary gland pathology and in growth of transplantable murine mammary cancer. J. Exp. Clin. Cancer Res. 2000, 19, 383–390.
- Watanabe, M.; Maemura, K.; Oki, K.; Shiraishi, N.; Shibayama, Y.; Katsu, K. Gamma-aminobutyric acid (GABA) and cell proliferation: Focus on cancer cells. Histol. Histopathol. 2006, 21, 1135–1141.
- Fasoulakis, Z.; Kolios, G.; Papamanolis, V.; Kontomanolis, E.N. Interleukins Associated with Breast Cancer. Cureus 2018, 10, e3549.
- Dmitrieva, O.S.; Shilovskiy, I.; Khaitov, M.; Grivennikov, S.I. Interleukins 1 and 6 as main mediators of inflammation and cancer. Biochemistry 2016, 81, 80–90.
- Anestakis, D.; Petanidis, S.; Kalyvas, S.; Nday, C.M.; Tsave, O.; Kioseoglou, E.; Salifoglou, A. Mechanisms and Applications of Interleukins in Cancer Immunotherapy. Int. J. Mol. Sci. 2015, 16, 1691–1710.
- Song, K.; Cai, X.; Dong, Y.; Wu, H.; Wei, Y.; Shankavaram, U.T.; Cui, K.; Lee, Y.; Zhu, B.; Bhattacharjee, S.; et al. Epsins 1 and 2 promote NEMO linear ubiquitination via LUBAC to drive breast cancer development. J. Clin. Investig. 2021, 131, e129374.
- Chen, H.; Fre, S.; Slepnev, V.I.; Capua, M.R.; Takei, K.; Butler, M.H.; Di Fiore, P.P.; De Camilli, P. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature 1998, 394, 793–797.
- Ko, G.; Paradise, S.; Chen, H.; Graham, M.; Vecchi, M.; Bianchi, F.; Cremona, O.; Di Fiore, P.P.; De Camilli, P. Selective high-level expression of epsin 3 in gastric parietal cells, where it is localized at endocytic sites of apical canaliculi. Proc. Natl. Acad. Sci. USA 2010, 107, 21511–21516.
- Spradling, K.D.; McDaniel, A.E.; Lohi, J.; Pilcher, B.K. Epsin 3 Is a Novel Extracellular Matrix-induced Transcript Specific to Wounded Epithelia. J. Biol. Chem. 2001, 276, 29257–29267.
- Bhattacharjee, S.; Lee, Y.; Zhu, B.; Wu, H.; Chen, Y.; Chen, H. Epsins in vascular development, function and disease. Cell. Mol. Life Sci. 2020, 78, 833–842.
- Windler, S.L.; Bilder, D. Endocytic Internalization Routes Required for Delta/Notch Signaling. Curr. Biol. 2010, 20, 538–543.
- Aguilar, R.C.; Longhi, S.A.; Shaw, J.D.; Yeh, L.-Y.; Kim, S.; Schon, A.; Freire, E.; Hsu, A.; McCormick, W.K.; Watson, H.A.; et al. Epsin N-terminal homology domains perform an essential function regulating Cdc42 through binding Cdc42 GTPase-activating proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 4116–4121.
- Rahman, H.A.; Wu, H.; Dong, Y.; Pasula, S.; Wen, A.; Sun, Y.; Brophy, M.L.; Tessneer, K.L.; Cai, X.; McManus, J.; et al. Selective Targeting of a Novel Epsin–VEGFR2 Interaction Promotes VEGF-Mediated Angiogenesis. Circ. Res. 2016, 118, 957–969.
- Liu, X.; Pasula, S.; Song, H.; Tessneer, K.L.; Dong, Y.; Hahn, S.; Yago, T.; Brophy, M.L.; Chang, B.; Cai, X.; et al. Temporal and spatial regulation of epsin abundance and VEGFR3 signaling are required for lymphatic valve formation and function. Sci. Signal. 2014, 7, ra97.
- Coon, B.G.; Burgner, J.; Camonis, J.H.; Aguilar, R.C. The Epsin Family of Endocytic Adaptors Promotes Fibrosarcoma Migration and Invasion. J. Biol. Chem. 2010, 285, 33073–33081.
- Coon, B.G.; DiRenzo, D.M.; Konieczny, S.F.; Aguilar, R.C. Epsins’ novel role in cancer cell invasion. Commun. Integr. Biol. 2011, 4, 95–97.
- Wang, Y.; Song, W.; Kan, P.; Huang, C.; Ma, Z.; Wu, Q.; Yao, X.; Zhang, B. Overexpression of Epsin 3 enhances migration and invasion of glioma cells by inducing epithelialmesenchymal transition. Oncol. Rep. 2018, 40, 3049–3059.
- Sloane, B.F.; Rozhin, J.; Johnson, K.; Taylor, H.; Crissman, J.D.; Honn, K.V. Cathepsin B: Association with plasma membrane in metastatic tumors. Proc. Natl. Acad. Sci. USA 1986, 83, 2483–2487.
- Brix, K. Lysosomal Proteases: Revival of the Sleeping Beauty. In Madame Curie Bioscience Database (Formerly, Eurekah Bioscience Database); Saftig, P., Ed.; Landes Bioscience: Austin, TX, USA, 2005.
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2011, 1824, 68–88.
- Pu, J.; Guardia, C.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339.
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431.
- Joyce, J.A.; Baruch, A.; Chehade, K.; Meyer-Morse, N.; Giraudo, E.; Tsai, F.-Y.; Greenbaum, D.C.; Hager, J.H.; Bogyo, M.; Hanahan, D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 2004, 5, 443–453.
- Wang, B.; Sun, J.; Kitamoto, S.; Yang, M.; Grubb, A.; Chapman, H.A.; Kalluri, R.; Shi, G.-P. Cathepsin S Controls Angiogenesis and Tumor Growth via Matrix-derived Angiogenic Factors. J. Biol. Chem. 2006, 281, 6020–6029.
- Burden, R.E.; Gormley, J.A.; Jaquin, T.J.; Small, D.; Quinn, D.J.; Hegarty, S.M.; Ward, C.; Walker, B.; Johnston, J.A.; Olwill, S.A.; et al. Antibody-Mediated Inhibition of Cathepsin S Blocks Colorectal Tumor Invasion and Angiogenesis. Clin. Cancer Res. 2009, 15, 6042–6051.
- Urbich, C.; Heeschen, C.; Aicher, A.; Sasaki, K.-I.; Bruhl, T.; Farhadi, M.R.; Vajkoczy, P.; Hofmann, W.K.; Peters, C.; Pennacchio, L.; et al. Cathepsin L is required for endothelial progenitor cell–induced neovascularization. Nat. Med. 2005, 11, 206–213.
- Yang, Z.; Cox, J.L. Cathepsin L increases invasion and migration of B16 melanoma. Cancer Cell Int. 2007, 7, 8.
- Rousselet, N.; Mills, L.; Jean, D.; Tellez, C.; Bar-Eli, M.; Frade, R. Inhibition of Tumorigenicity and Metastasis of Human Melanoma Cells by Anti-Cathepsin L Single Chain Variable Fragment. Cancer Res. 2004, 64, 146–151.
- Gocheva, V.; Joyce, J.A. Cysteine Cathepsins and the Cutting Edge of Cancer Invasion. Cell Cycle 2007, 6, 60–64.
- S Sudhan, D.R.; Siemann, D.W. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin. Exp. Metastasis 2013, 30, 891–902.
- Bratovš, A.; Kramer, L.; Mikhaylov, G.; Vasiljeva, O.; Turk, B. Stefin A-functionalized liposomes as a system for cathepsins S and L-targeted drug delivery. Biochimie 2019, 166, 94–102.
- Pedersen, M.; Larsen, A.; Stoltenberg, M.; Penkowa, M. The role of metallothionein in oncogenesis and cancer prognosis. Prog. Histochem. Cytochem. 2009, 44, 29–64.
- Thirumoorthy, N. Metallothionein: An overview. World J. Gastroenterol. 2007, 13, 993–996.
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 2018, 11, 1–20.
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Experientia 2002, 59, 627–647.
- Krężel, A.; Maret, W. The Functions of Metamorphic Metallothioneins in Zinc and Copper Metabolism. Int. J. Mol. Sci. 2017, 18, 1237.
- Kumari, M.R.; Hiramatsu, M.; Ebadi, M. Free radical scavenging actions of metallothionein isoforms I and II. Free Radic. Res. 1998, 29, 93–101.
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The Role of Metallothionein in Oxidative Stress. Int. J. Mol. Sci. 2013, 14, 6044–6066.
- Klaassen, C.D.; Liu, J.; Diwan, B.A. Metallothionein protection of cadmium toxicity. Toxicol. Appl. Pharmacol. 2009, 238, 215–220.
- Arriaga, J.M.; Levy, E.M.; Bravo, A.I.; Bayo, S.M.; Amat, M.; Aris, M.; Hannois, A.; Bruno, L.; Roberti, M.P.; Loria, F.S.; et al. Metallothionein expression in colorectal cancer: Relevance of different isoforms for tumor progression and patient survival. Hum. Pathol. 2012, 43, 197–208.
- Demidenko, R.; Daniunaite, K.; Bakavicius, A.; Sabaliauskaite, R.; Skeberdyte, A.; Petroska, D.; Laurinavicius, A.; Jankevicius, F.; Lazutka, J.R.; Jarmalaite, S. Decreased expression of MT1E is a potential biomarker of prostate cancer progression. Oncotarget 2017, 8, 61709–61718.
- Zheng, Y.; Jiang, L.; Hu, Y.; Xiao, C.; Xu, N.; Zhou, J.; Zhou, X. Metallothionein 1H (MT1H) functions as a tumor suppressor in hepatocellular carcinoma through regulating Wnt/β-catenin signaling pathway. BMC Cancer 2017, 17, 1–11.
- Gomulkiewicz, A.; Podhorska-Okolow, M.; Szulc, R.; Smorag, Z.; Wojnar, A.; Zabel, M.; Dzięgiel, P. Correlation between metallothionein (MT) expression and selected prognostic factors in ductal breast cancers. Folia Histochem. Cytobiol. 2010, 48, 242–248.
- Hengstler, J.; Pilch, H.; Schmidt, M.; Dahlenburg, H.; Schiffer, I.; Oesch, F.; Knapstein, P.; Kaina, B.; Tanner, B. Metallothionein expression in ovarian cancer in relation to histopathological parameters and molecular markers of prognosis. Int. J. Cancer 2001, 95, 121–127.
- Wülfing, C.; Van Ahlen, H.; Eltze, E.; Piechota, H.; Hertle, L.; Schmid, K.-W. Metallothionein in bladder cancer: Correlation of overexpression with poor outcome after chemotherapy. World J. Urol. 2007, 25, 199–205.
- Jayasurya, A.; Bay, B.; Yap, W.; Tan, N.; Tan, B. Proliferative potential in nasopharyngeal carcinoma: Correlations with metallothionein expression and tissue zinc levels. Carcinogenesis 2000, 21, 1809–1812.
- Weinlich, G.; Eisendle, K.; Hassler, E.; Baltaci, M.; O Fritsch, P.; Zelger, B. Metallothionein – overexpression as a highly significant prognostic factor in melanoma: A prospective study on 1270 patients. Br. J. Cancer 2006, 94, 835–841.
- Werynska, B.; Pula, B.; Muszczynska-Bernhard, B.; Piotrowska, A.; Jethon, A.; Podhorska-Okolow, M.; Dziegiel, P.; Jankowska, R. Correlation between expression of metallothionein and expression of Ki-67 and MCM-2 proliferation markers in non-small cell lung cancer. Anticancer. Res. 2011, 31, 2833–2839.
- Jin, R.; Chow, V.T.-K.; Tan, P.-H.; Dheen, S.T.; Duan, W.; Bay, B.-H. Metallothionein 2A expression is associated with cell proliferation in breast cancer. Carcinogenesis 2002, 23, 81–86.
- Jin, R.; Bay, B.-H.; Chow, V.T.-K.; Tan, P.-H. Metallothionein 1F mRNA expression correlates with histological grade in breast carcinoma. Breast Cancer Res. Treat. 2001, 66, 265–272.
- Wierzowiecka, B.; Gomulkiewicz, A.; Cwynar-Zając, L.; Olbromski, M.; Grzegrzolka, J.; Kobierzycki, C.; Podhorska-Okolow, M.; Dziegiel, P. Expression of Metallothionein and Vascular Endothelial Growth Factor Isoforms in Breast Cancer Cells. In Vivo 2016, 30, 271–278.