In a traumatically injured brain, the cerebral microdialysis technique allows continuous sampling of fluid from the brain’s extracellular space. The retrieved brain fluid contains useful metabolites that indicate the brain’s energy state. Assessment of these metabolites along with other parameters, such as intracranial pressure, brain tissue oxygenation, and cerebral perfusion pressure, may help inform clinical decision making, guide medical treatments, and aid in the prognostication of patient outcomes. Currently, brain metabolites are assayed on bedside analysers and results can only be achieved hourly. This is a major drawback because critical information within each hour is lost. To address this, recent advances have focussed on developing biosensing techniques for integration with microdialysis to achieve continuous online monitoring.
- biosensors
- traumatic brain injury
- neurochemistry
- cerebral microdialysis
- cerebral metabolism
1. Introduction
2. Importance of Monitoring Brain Metabolism for TBI
2.1. Traumatic Brain Injury (TBI)
TBI is an insult to the brain due to an external force causing damage to the brain’s structure and function [9]. It represents mild, moderate, and severe effects of physical assault to the brain that may cause sequential, primary, or secondary ramifications. Thus, TBI can be classed into a primary mechanical injury, which is non-amenable to medical treatment and a delayed secondary brain injury involving various changes at the cellular and molecular level that could in theory be counteracted. The sudden mechanical injury is a widespread tearing, shearing, or stretching of axons, referred to as axonal injury, as well as contusions, hemorrhages, and lacerations [10]. Delayed secondary injury evolves hours or days after the initial primary mechanical trauma due to neuronal and glial dysfunction, neuroinflammation, cerebral oedema, and metabolic changes. Consequently, this leads to various physiologic alterations including hypoperfusion, blood–brain barrier (BBB) disruption, oxidative injury, and mitochondrial dysfunction [11,12,13,14,15,16][11][12][13][14][15][16]. This complexity of secondary brain injury poses diagnostic and therapeutic challenges; therefore, additional invasive monitoring interventions are required during neurocritical care of severe TBI patients. These additional metrics may provide insights into brain tissue oxygenation, intracranial pressure (ICP), cerebral perfusion pressure (CPP), electrophysiology, and local brain metabolism [11]. Abnormal local brain metabolism is linked to poor patient clinical outcomes, thus interrogating the brain to monitor its chemistry is necessary [17].2.2. Cerebral Metabolism
Evidence shows that cerebral metabolism is disturbed following TBI, although the exact mechanisms are incompletely understood due to the complex and heterogeneous nature of TBI [7]. The brain uses glucose as a preferred substrate for energy consumption, so the regulation of cerebral glucose metabolism is crucial. Oxidative metabolism of glucose provides most of the ATPs utilised by the brain. However, biosynthetic routes that branch from the glycolytic pathway and the tricarboxylic acid (TCA) cycle and other pathways including the pentose phosphate shunt, glucose storage as glycogen, and the malate-aspartate shuttle all have significant roles [18]. Understanding of the altered cerebral metabolism is incomplete without knowledge of the glucose metabolism in an uninjured brain. Figure 1 presents the major energy pathways in the brain.
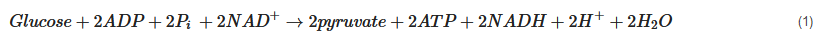 Pyruvate, the end-product of glycolysis, can enter mitochondria where it is converted to acetyl-CoA by pyruvate dehydrogenase. Acetyl-CoA is further metabolised in the tricarboxylic acid (TCA) cycle in mitochondria. The sum of all the reactions in the TCA cycle is presented in Equation (2).
Pyruvate, the end-product of glycolysis, can enter mitochondria where it is converted to acetyl-CoA by pyruvate dehydrogenase. Acetyl-CoA is further metabolised in the tricarboxylic acid (TCA) cycle in mitochondria. The sum of all the reactions in the TCA cycle is presented in Equation (2).
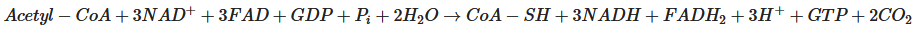 Subsequently, NADH and FADH2 are utilised by the mitochondrial electron transport chain (by Complexes I and II, respectively); electrons are transferred to complexes III and IV, where O2 is the terminal electron acceptor on Complex IV, followed by ATP synthesis by ATP synthase (also termed Complex V) [20]. The yield per molecule of glucose metabolised fully to CO2 (by combined glycolysis, NADH shuttling, and mitochondrial respiration) is theoretically 36–38 ATP molecules. However, the actual yield is considered somewhat lower [21,22][21][22].
Subsequently, NADH and FADH2 are utilised by the mitochondrial electron transport chain (by Complexes I and II, respectively); electrons are transferred to complexes III and IV, where O2 is the terminal electron acceptor on Complex IV, followed by ATP synthesis by ATP synthase (also termed Complex V) [20]. The yield per molecule of glucose metabolised fully to CO2 (by combined glycolysis, NADH shuttling, and mitochondrial respiration) is theoretically 36–38 ATP molecules. However, the actual yield is considered somewhat lower [21,22][21][22].
2.3. Altered Cerebral Metabolism Due to TBI
Imbalance in cerebral glucose metabolism following TBI is well-documented [7,23,24][7][23][24] and attributable, at least partly, to the altered ATP production in the brain’s major energy pathways. Due to a high energy demand following TBI, abnormally low levels (<0.8 mM) of extracellular glucose occur [17], possibly because of upregulated glucose uptake by neurones and glia. Conversely, neurones and glia may sometimes be too damaged to take up glucose from extracellular fluid, leading to hypometabolism characterised by abnormally high extracellular glucose. Thus, there is an optimum extracellular glucose range, although there is insufficient evidence to define this exactly [25]. Extracellular lactate can also be utilised as an alternative fuel [24]. 13C-labelled microdialysis studies have demonstrated that the traumatically injured brain uses lactate via the TCA cycle [24,26][24][26]. Another 13C-labelled microdialysis study found lactate production from 1,2-13C2 glucose via glycolysis and to a lesser extent via PPP [23]. Lactate was also identified as a spin-out product (cataplerosis) from the TCA cycle in 13C-labelled microdialysis studies using 2,3-13C2 succinate as a substrate [11,23][11][23]. A persistent high lactate/pyruvate ratio (LPR) (LPR > 25 or >40) indicates metabolic dysfunction or crisis. In a microdialysis study of 233 TBI patients, acute-phase LPR > 25 was associated with poor clinical outcomes 6 months later [17]. High LPR, despite seemingly adequate oxygen and glucose delivery to brain tissues, is regarded as indicating mitochondrial dysfunction [11,27][11][27]. The concentrations of lactate and pyruvate and their ratio (LPR) provide useful information about the cellular redox state in the region of interest. The extracellular LPR is thought to reflect the LPR in the cytoplasm—itself in equilibrium with cytoplasmic NADH/NAD+ ratio [28]. Glucose, lactate, pyruvate, and LPR were cited as the most clinically relevant biomarkers in a consensus statement from the 2014 International Microdialysis Forum [25]. Timely assessment of these is, therefore, essential in the early detection of secondary brain injury allowing prompt interventions. Table 1 summarises neuroprotective interventions for altered neurochemistry.|
Intervention |
Effect |
References |
||||
|---|---|---|---|---|---|---|
|
Glucose/insulin |
↑↓ glucose, ↑↓ LPR |
|||||
In-vitro |
Measured 0–100 μM glucose concentration, with 25 μM increments, in a microdialysate stream. |
Hyperoxia |
↑ PBtO2, variable ↓ LPR |
|||
|
Pagkalos et al. (2018) [46 | [ | 36] |
||||
] |
Enzymatic-electrochemical |
In-vitro |
Measured 0–50 μM lactate concentrations with 12.5 μM increments using enzymatic based sensor with LoD range 2.5 to 9.5 nM, in a microdialysate stream. |
Hyperventilation |
||
|
Tageldeen et al. (2020) [47] |
↓ glucose |
Enzymatic-electrochemical |
||||
In-vitro |
Measured 0–1 mM glucose and lactate, changing concentrations. LoDs of 0.85 and 1.3 μM for glucose and lactate, respectively, in a microdialysate stream. |
Mannitol |
||||
|
Robbins et al. (2019) [ |
↓ LPR |
|||||
48] |
Enzymatic-electrochemical |
In-vivo (rats) |
Reported progressive decrease in glucose in microdialysates from a cortical impact injury. |
Decompressive craniotomy |
↓ LPR |
|
|
Rogers et al. (2017) [50] | [43 |
Enzymatic-electrochemical |
||||
|
Therapeutic (induced) hypothermia |
↓ glucose, ↓ lactate |
3. Review of Sensor Technologies for Brain Metabolism
In selecting scholarly articles to discuss here, emphasis was placed on work published in the areas of biosensors, TBI, cerebral metabolism, and microdialysis in the last 10 years. Table 2 summarises some of the most relevant articles discussed here. Those biosensors are in the research and development phases and are not approved for routine clinical use.|
Study |
Sensor Type |
Setting |
Comments |
|---|---|---|---|
|
Papadimitriou et al. (2016) [45] |
Enzymatic-electrochemical |
||
In-vivo (human) | |||
Continuous online microdialysis measurements in TBI patients; monitoring duration > 6 h; glucose, lactate, and K | + levels in spreading depolarisation (K+ was measured by an ion-selective electrode). |
||
|
Gowers et al. (2019) [52] |
Enzymatic-electrochemical |
In-vivo (human) |
Detected a sudden surge of lactate levels during continuous online dialysate measurements in TBI patients. |
|
Gifford et al. (2021) [51] |
Enzymatic-electrochemical |
In-vivo (human) |
Reported declining glucose levels in 3 TBI patients, and persistent low glucose in 1 TBI patient, in dexamethasone-enhanced continuous online microdialysis. |
|
Alimagham et al. (2021) [49] |
Optical (mid-IR) |
Ex-vivo (human) |
Microdialysate measurements from TBI patients, offline. LoDs of 0.5, 0.2, and 0.1 mM for glucose, lactate, and pyruvate respectively. Quantification of brain metabolites was compared with a conventional enzymatic-colorimetric microdialysis analyser (ISCUSflex). |
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048.
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097.
- Barlow, K.M. Traumatic brain injury. Handb. Clin. Neurol. 2013, 112, 891–904.
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths-United States, 2007 and 2013. MMWR Surveill. Summ. 2017, 66, 1–16.
- Vidgeon, S.D.; Strong, A.J. Multimodal Cerebral Monitoring in Traumatic Brain Injury. J. Intensive Care Soc. 2011, 12, 126–133.
- Wijayatilake, D.S.; Shepherd, S.J. What’s new in the management of traumatic brain injury on neuro ICU? Curr. Opin. Anaesthesiol. 2014, 27, 459–464.
- Carpenter, K.L.; Jalloh, I.; Gallagher, C.N.; Grice, P.; Howe, D.J.; Mason, A.; Timofeev, I.; Helmy, A.; Murphy, M.P.; Menon, D.K.; et al. 13C-labelled microdialysis studies of cerebral metabolism in TBI patients. Eur. J. Pharm. Sci. 2014, 57, 87–97.
- Helmy, A.; Carpenter, K.L.; Hutchinson, P.J. Microdialysis in the human brain and its potential role in the development and clinical assessment of drugs. Curr. Med. Chem. 2007, 14, 1525–1537.
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Position statement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640.
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528.
- Khellaf, A.; Khan, D.Z.; Helmy, A. Recent advances in traumatic brain injury. J. Neurol. 2019, 266, 2878–2889.
- Chamoun, R.; Suki, D.; Gopinath, S.P.; Goodman, J.C.; Robertson, C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J. Neurosurg. 2010, 113, 564–570.
- Dyhrfort, P.; Shen, Q.; Clausen, F.; Thulin, M.; Enblad, P.; Kamali-Moghaddam, M.; Lewén, A.; Hillered, L. Monitoring of Protein Biomarkers of Inflammation in Human Traumatic Brain Injury Using Microdialysis and Proximity Extension Assay Technology in Neurointensive Care. J. Neurotrauma 2019, 36, 2872–2885.
- Lassarén, P.; Lindblad, C.; Frostell, A.; Carpenter, K.L.H.; Guilfoyle, M.R.; Hutchinson, P.J.A.; Helmy, A.; Thelin, E.P. Systemic inflammation alters the neuroinflammatory response: A prospective clinical trial in traumatic brain injury. J. Neuroinflamm. 2021, 18, 221.
- Di, X.; Lyeth, B.G.; Hamm, R.J.; Bullock, M.R. Voltage-dependent Na+/K+ ion channel blockade fails to ameliorate behavioral deficits after traumatic brain injury in the rat. J. Neurotrauma 1996, 13, 497–504.
- Guilfoyle, M.R.; Carpenter, K.L.; Helmy, A.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Matrix Metalloproteinase Expression in Contusional Traumatic Brain Injury: A Paired Microdialysis Study. J. Neurotrauma 2015, 32, 1553–1559.
- Timofeev, I.; Carpenter, K.L.H.; Nortje, J.; Al-Rawi, P.G.; O’Connell, M.T.; Czosnyka, M.; Smielewski, P.; Pickard, J.D.; Menon, D.K.; Kirkpatrick, P.J.; et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: A microdialysis study of 223 patients. Brain 2011, 134, 484–494.
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045.
- McKenna, M.C.; Waagepetersen, H.S.; Schousboe, A.; Sonnewald, U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: Current evidence and pharmacological tools. Biochem. Pharm. 2006, 71, 399–407.
- Rich, P.R.; Maréchal, A. The mitochondrial respiratory chain. Essays Biochem. 2010, 47, 1–23.
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J., Jr.; Stryer, L. Biochemistry, 9th ed.; McMillan Learning: New York, NY, USA, 2019.
- Lodish, H.F.; Berk, A.; Kaiser, C.; Krieger, M.; Scott, M.P.; Bretscher, A.; Ploegh, H.L.; Matsudaira, P.T. Molecular Cell Biology, 6th ed.; W.H. Freeman: New York, NY, USA, 2008.
- Jalloh, I.; Carpenter, K.L.H.; Grice, P.; Howe, D.J.; Mason, A.; Gallagher, C.N.; Helmy, A.; Murphy, M.P.; Menon, D.K.; Carpenter, T.A.; et al. Glycolysis and the Pentose Phosphate Pathway after Human Traumatic Brain Injury: Microdialysis Studies Using 1,2-13C2 Glucose. J. Cereb. Blood Flow Metab. 2015, 35, 111–120.
- Jalloh, I.; Helmy, A.; Howe, D.J.; Shannon, R.J.; Grice, P.; Mason, A.; Gallagher, C.N.; Murphy, M.P.; Pickard, J.D.; Menon, D.K.; et al. A Comparison of Oxidative Lactate Metabolism in Traumatically Injured Brain and Control Brain. J. Neurotrauma 2018, 35, 2025–2035.
- Hutchinson, P.J.; Jalloh, I.; Helmy, A.; Carpenter, K.L.; Rostami, E.; Bellander, B.M.; Boutelle, M.G.; Chen, J.W.; Claassen, J.; Dahyot-Fizelier, C.; et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med. 2015, 41, 1517–1528.
- Gallagher, C.N.; Carpenter, K.L.; Grice, P.; Howe, D.J.; Mason, A.; Timofeev, I.; Menon, D.K.; Kirkpatrick, P.J.; Pickard, J.D.; Sutherland, G.R.; et al. The human brain utilizes lactate via the tricarboxylic acid cycle: A 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain 2009, 132, 2839–2849.
- Guilfoyle, M.R.; Helmy, A.; Donnelly, J.; Stovell, M.G.; Timofeev, I.; Pickard, J.D.; Czosnyka, M.; Smielewski, P.; Menon, D.K.; Carpenter, K.L.H.; et al. Characterising the dynamics of cerebral metabolic dysfunction following traumatic brain injury: A microdialysis study in 619 patients. PLoS ONE 2021, 16, e0260291.
- Williamson, D.H.; Lund, P.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 1967, 103, 514–527.
- Vespa, P.; Boonyaputthikul, R.; McArthur, D.L.; Miller, C.; Etchepare, M.; Bergsneider, M.; Glenn, T.; Martin, N.; Hovda, D. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit. Care Med. 2006, 34, 850–856.
- Vespa, P.; McArthur, D.L.; Stein, N.; Huang, S.C.; Shao, W.; Filippou, M.; Etchepare, M.; Glenn, T.; Hovda, D.A. Tight glycemic control increases metabolic distress in traumatic brain injury: A randomized controlled within-subjects trial. Crit. Care Med. 2012, 40, 1923–1929.
- Oddo, M.; Schmidt, J.M.; Carrera, E.; Badjatia, N.; Connolly, E.S.; Presciutti, M.; Ostapkovich, N.D.; Levine, J.M.; Le Roux, P.; Mayer, S.A. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: A microdialysis study. Crit. Care Med. 2008, 36, 3233–3238.
- Helbok, R.; Schmidt, J.M.; Kurtz, P.; Hanafy, K.A.; Fernandez, L.; Stuart, R.M.; Presciutti, M.; Ostapkovich, N.D.; Connolly, E.S.; Lee, K.; et al. Systemic glucose and brain energy metabolism after subarachnoid hemorrhage. Neurocrit. Care 2010, 12, 317–323.
- Rostami, E.; Bellander, B.M. Monitoring of glucose in brain, adipose tissue, and peripheral blood in patients with traumatic brain injury: A microdialysis study. J. Diabetes Sci. Technol. 2011, 5, 596–604.
- Tolias, C.M.; Reinert, M.; Seiler, R.; Gilman, C.; Scharf, A.; Bullock, M.R. Normobaric hyperoxia--induced improvement in cerebral metabolism and reduction in intracranial pressure in patients with severe head injury: A prospective historical cohort-matched study. J. Neurosurg. 2004, 101, 435–444.
- Nortje, J.; Coles, J.P.; Timofeev, I.; Fryer, T.D.; Aigbirhio, F.I.; Smielewski, P.; Outtrim, J.G.; Chatfield, D.A.; Pickard, J.D.; Hutchinson, P.J.; et al. Effect of hyperoxia on regional oxygenation and metabolism after severe traumatic brain injury: Preliminary findings. Crit. Care Med. 2008, 36, 273–281.
- Rockswold, S.B.; Rockswold, G.L.; Zaun, D.A.; Liu, J. A prospective, randomized Phase II clinical trial to evaluate the effect of combined hyperbaric and normobaric hyperoxia on cerebral metabolism, intracranial pressure, oxygen toxicity, and clinical outcome in severe traumatic brain injury. J. Neurosurg. 2013, 118, 1317–1328.
- Marion, D.W.; Puccio, A.; Wisniewski, S.R.; Kochanek, P.; Dixon, C.E.; Bullian, L.; Carlier, P. Effect of hyperventilation on extracellular concentrations of glutamate, lactate, pyruvate, and local cerebral blood flow in patients with severe traumatic brain injury. Crit. Care Med. 2002, 30, 2619–2625.
- Hutchinson, P.J.; Gupta, A.K.; Fryer, T.F.; Al-Rawi, P.G.; Chatfield, D.A.; Coles, J.P.; O’Connell, M.T.; Kett-White, R.; Minhas, P.S.; Aigbirhio, F.I.; et al. Correlation between Cerebral Blood Flow, Substrate Delivery, and Metabolism in Head Injury: A Combined Microdialysis and Triple Oxygen Positron Emission Tomography Study. J. Cereb. Blood Flow Metab. 2002, 22, 735–745.
- Sakowitz, O.W.; Stover, J.F.; Sarrafzadeh, A.S.; Unterberg, A.W.; Kiening, K.L. Effects of mannitol bolus administration on intracranial pressure, cerebral extracellular metabolites, and tissue oxygenation in severely head-injured patients. J. Trauma 2007, 62, 292–298.
- Chiu, C.; Xian, W.; Moss, C.F. Flying in silence: Echolocating bats cease vocalizing to avoid sonar jamming. Proc. Natl. Acad. Sci. USA 2008, 105, 13116–13121.
- Soukup, J.; Zauner, A.; Doppenberg, E.M.; Menzel, M.; Gilman, C.; Bullock, R.; Young, H.F. Relationship between brain temperature, brain chemistry and oxygen delivery after severe human head injury: The effect of mild hypothermia. Neurol. Res. 2002, 24, 161–168.
- Berger, C.; Schäbitz, W.R.; Georgiadis, D.; Steiner, T.; Aschoff, A.; Schwab, S. Effects of hypothermia on excitatory amino acids and metabolism in stroke patients: A microdialysis study. Stroke 2002, 33, 519–524.
- Ho, C.L.; Wang, C.M.; Lee, K.K.; Ng, I.; Ang, B.T. Cerebral oxygenation, vascular reactivity, and neurochemistry following decompressive craniectomy for severe traumatic brain injury. J. Neurosurg. 2008, 108, 943–949.
- Nagel, A.; Graetz, D.; Schink, T.; Frieler, K.; Sakowitz, O.; Vajkoczy, P.; Sarrafzadeh, A. Relevance of intracranial hypertension for cerebral metabolism in aneurysmal subarachnoid hemorrhage. Clinical article. J. Neurosurg. 2009, 111, 94–101.
- Papadimitriou, K.I.; Wang, C.; Rogers, M.L.; Gowers, S.A.N.; Leong, C.L.; Boutelle, M.G.; Drakakis, E.M. High-Performance Bioinstrumentation for Real-Time Neuroelectrochemical Traumatic Brain Injury Monitoring. Front. Hum. Neurosci. 2016, 10, 212.
- Pagkalos, I.; Rogers, M.; Boutelle, M.; Drakakis, E. A Higà Performance Application Specific Integrated Circuit for Electrical and Neurochemical Traumatic Brain Injury Monitoring. Chemphyschem 2018, 19, 1215–1225.
- Tageldeen, M.K.; Gowers, S.A.N.; Leong, C.L.; Boutelle, M.G.; Drakakis, E.M. Traumatic brain injury neuroelectrochemical monitoring: Behind-the-ear micro-instrument and cloud application. J. Neuroeng. Rehabil. 2020, 17, 114.
- Robbins, E.M.; Jaquins-Gerstl, A.; Fine, D.F.; Leong, C.L.; Dixon, C.E.; Wagner, A.K.; Boutelle, M.G.; Michael, A.C. Extended (10-Day) Real-Time Monitoring by Dexamethasone-Enhanced Microdialysis in the Injured Rat Cortex. ACS Chem. Neurosci. 2019, 10, 3521–3531.
- Alimagham, F.C.; Hutter, D.; Marco-García, N.; Gould, E.; Highland, V.H.; Huefner, A.; Giorgi-Coll, S.; Killen, M.J.; Zakrzewska, A.P.; Elliott, S.R.; et al. Cerebral Microdialysate Metabolite Monitoring using Mid-infrared Spectroscopy. Anal. Chem. 2021, 93, 11929–11936.
- Rogers, M.L.; Leong, C.L.; Gowers, S.A.; Samper, I.C.; Jewell, S.L.; Khan, A.; McCarthy, L.; Pahl, C.; Tolias, C.M.; Walsh, D.C.; et al. Simultaneous monitoring of potassium, glucose and lactate during spreading depolarization in the injured human brain–Proof of principle of a novel real-time neurochemical analysis system, continuous online microdialysis. J. Cereb. Blood Flow Metab. 2017, 37, 1883–1895.
- Gifford, E.K.; Robbins, E.M.; Jaquins-Gerstl, A.; Rerick, M.T.; Nwachuku, E.L.; Weber, S.G.; Boutelle, M.G.; Okonkwo, D.O.; Puccio, A.M.; Michael, A.C. Validation of Dexamethasone-Enhanced Continuous-Online Microdialysis for Monitoring Glucose for 10 Days after Brain Injury. ACS Chem. Neurosci. 2021, 12, 3588–3597.
- Hutchinson, P.J. Microdialysis in traumatic brain injury--methodology and pathophysiology. Acta Neurochir. Suppl. 2005, 95, 441–445.