In a traumatically injured brain, the cerebral microdialysis technique allows continuous sampling of fluid from the brain’s extracellular space. The retrieved brain fluid contains useful metabolites that indicate the brain’s energy state. Assessment of these metabolites along with other parameters, such as intracranial pressure, brain tissue oxygenation, and cerebral perfusion pressure, may help inform clinical decision making, guide medical treatments, and aid in the prognostication of patient outcomes. Currently, brain metabolites are assayed on bedside analysers and results can only be achieved hourly. This is a major drawback because critical information within each hour is lost. To address this, recent advances have focussed on developing biosensing techniques for integration with microdialysis to achieve continuous online monitoring.
- biosensors
- traumatic brain injury
- neurochemistry
- cerebral microdialysis
- cerebral metabolism
1. Introduction
2. Importance of Monitoring Brain Metabolism for TBI
2.1. Traumatic Brain Injury (TBI)
TBI is an insult to the brain due to an external force causing damage to the brain’s structure and function [9]. It represents mild, moderate, and severe effects of physical assault to the brain that may cause sequential, primary, or secondary ramifications. Thus, TBI can be classed into a primary mechanical injury, which is non-amenable to medical treatment and a delayed secondary brain injury involving various changes at the cellular and molecular level that could in theory be counteracted. The sudden mechanical injury is a widespread tearing, shearing, or stretching of axons, referred to as axonal injury, as well as contusions, hemorrhages, and lacerations [10]. Delayed secondary injury evolves hours or days after the initial primary mechanical trauma due to neuronal and glial dysfunction, neuroinflammation, cerebral oedema, and metabolic changes. Consequently, this leads to various physiologic alterations including hypoperfusion, blood–brain barrier (BBB) disruption, oxidative injury, and mitochondrial dysfunction [11][12][13][14][15][16][11,12,13,14,15,16]. This complexity of secondary brain injury poses diagnostic and therapeutic challenges; therefore, additional invasive monitoring interventions are required during neurocritical care of severe TBI patients. These additional metrics may provide insights into brain tissue oxygenation, intracranial pressure (ICP), cerebral perfusion pressure (CPP), electrophysiology, and local brain metabolism [11]. Abnormal local brain metabolism is linked to poor patient clinical outcomes, thus interrogating the brain to monitor its chemistry is necessary [17].2.2. Cerebral Metabolism
Evidence shows that cerebral metabolism is disturbed following TBI, although the exact mechanisms are incompletely understood due to the complex and heterogeneous nature of TBI [7]. The brain uses glucose as a preferred substrate for energy consumption, so the regulation of cerebral glucose metabolism is crucial. Oxidative metabolism of glucose provides most of the ATPs utilised by the brain. However, biosynthetic routes that branch from the glycolytic pathway and the tricarboxylic acid (TCA) cycle and other pathways including the pentose phosphate shunt, glucose storage as glycogen, and the malate-aspartate shuttle all have significant roles [18]. Understanding of the altered cerebral metabolism is incomplete without knowledge of the glucose metabolism in an uninjured brain. Figure 1 presents the major energy pathways in the brain.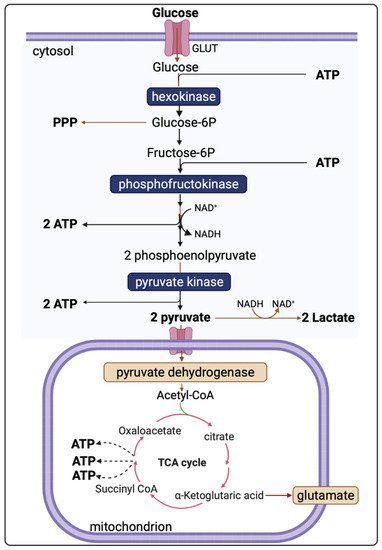
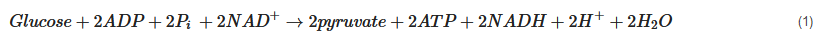 Pyruvate, the end-product of glycolysis, can enter mitochondria where it is converted to acetyl-CoA by pyruvate dehydrogenase. Acetyl-CoA is further metabolised in the tricarboxylic acid (TCA) cycle in mitochondria. The sum of all the reactions in the TCA cycle is presented in Equation (2).
Pyruvate, the end-product of glycolysis, can enter mitochondria where it is converted to acetyl-CoA by pyruvate dehydrogenase. Acetyl-CoA is further metabolised in the tricarboxylic acid (TCA) cycle in mitochondria. The sum of all the reactions in the TCA cycle is presented in Equation (2).
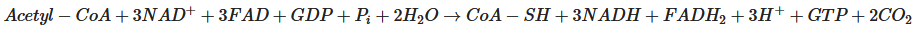 Subsequently, NADH and FADH2 are utilised by the mitochondrial electron transport chain (by Complexes I and II, respectively); electrons are transferred to complexes III and IV, where O2 is the terminal electron acceptor on Complex IV, followed by ATP synthesis by ATP synthase (also termed Complex V) [20]. The yield per molecule of glucose metabolised fully to CO2 (by combined glycolysis, NADH shuttling, and mitochondrial respiration) is theoretically 36–38 ATP molecules. However, the actual yield is considered somewhat lower [21][22][21,22].
Subsequently, NADH and FADH2 are utilised by the mitochondrial electron transport chain (by Complexes I and II, respectively); electrons are transferred to complexes III and IV, where O2 is the terminal electron acceptor on Complex IV, followed by ATP synthesis by ATP synthase (also termed Complex V) [20]. The yield per molecule of glucose metabolised fully to CO2 (by combined glycolysis, NADH shuttling, and mitochondrial respiration) is theoretically 36–38 ATP molecules. However, the actual yield is considered somewhat lower [21][22][21,22].
2.3. Altered Cerebral Metabolism Due to TBI
Imbalance in cerebral glucose metabolism following TBI is well-documented [7][23][24][7,23,24] and attributable, at least partly, to the altered ATP production in the brain’s major energy pathways. Due to a high energy demand following TBI, abnormally low levels (<0.8 mM) of extracellular glucose occur [17], possibly because of upregulated glucose uptake by neurones and glia. Conversely, neurones and glia may sometimes be too damaged to take up glucose from extracellular fluid, leading to hypometabolism characterised by abnormally high extracellular glucose. Thus, there is an optimum extracellular glucose range, although there is insufficient evidence to define this exactly [25]. Extracellular lactate can also be utilised as an alternative fuel [24]. 13C-labelled microdialysis studies have demonstrated that the traumatically injured brain uses lactate via the TCA cycle [24][26][24,26]. Another 13C-labelled microdialysis study found lactate production from 1,2-13C2 glucose via glycolysis and to a lesser extent via PPP [23]. Lactate was also identified as a spin-out product (cataplerosis) from the TCA cycle in 13C-labelled microdialysis studies using 2,3-13C2 succinate as a substrate [11][23][11,23]. A persistent high lactate/pyruvate ratio (LPR) (LPR > 25 or >40) indicates metabolic dysfunction or crisis. In a microdialysis study of 233 TBI patients, acute-phase LPR > 25 was associated with poor clinical outcomes 6 months later [17]. High LPR, despite seemingly adequate oxygen and glucose delivery to brain tissues, is regarded as indicating mitochondrial dysfunction [11][27][11,27]. The concentrations of lactate and pyruvate and their ratio (LPR) provide useful information about the cellular redox state in the region of interest. The extracellular LPR is thought to reflect the LPR in the cytoplasm—itself in equilibrium with cytoplasmic NADH/NAD+ ratio [28]. Glucose, lactate, pyruvate, and LPR were cited as the most clinically relevant biomarkers in a consensus statement from the 2014 International Microdialysis Forum [25]. Timely assessment of these is, therefore, essential in the early detection of secondary brain injury allowing prompt interventions. Table 1 summarises neuroprotective interventions for altered neurochemistry.Intervention | Effect | References | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Glucose/insulin | Papadimitriou et al. (2016) [45] | ↑↓ glucose, ↑↓ LPR | Enzymatic-electrochemical | ||||||||||||
In-vitro | Measured 0–100 μM glucose concentration, with 25 μM increments, in a microdialysate stream. | Hyperoxia | |||||||||||||
Pagkalos et al. (2018) [46] | ↑ PBtO2, variable ↓ LPR | ||||||||||||||
Enzymatic-electrochemical | In-vitro | Measured 0–50 μM lactate concentrations with 12.5 μM increments using enzymatic based sensor with LoD range 2.5 to 9.5 nM, in a microdialysate stream. | Hyperventilation | ↓ glucose | |||||||||||
Tageldeen et al. (2020) [47] | Enzymatic-electrochemical | ] | |||||||||||||
In-vitro | Measured 0–1 mM glucose and lactate, changing concentrations. LoDs of 0.85 and 1.3 μM for glucose and lactate, respectively, in a microdialysate stream. | Mannitol | |||||||||||||
Robbins et al. (2019) [48] | ↓ LPR | Enzymatic-electrochemical | |||||||||||||
In-vivo | (rats) | Reported progressive decrease in glucose in microdialysates from a cortical impact injury. | Decompressive craniotomy | ||||||||||||
Rogers et al. (2017) [50] | ↓ LPR | Enzymatic-electrochemical | |||||||||||||
Therapeutic (induced) hypothermia | ↓ glucose, ↓ lactate |
3. Review of Sensor Technologies for Brain Metabolism
In selecting scholarly articles to discuss here, emphasis was placed on work published in the areas of biosensors, TBI, cerebral metabolism, and microdialysis in the last 10 years. Table 2 summarises some of the most relevant articles discussed here. Those biosensors are in the research and development phases and are not approved for routine clinical use.Study | Sensor Type | Setting | Comments | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
In-vivo (human) | |||||||||||||
Continuous online microdialysis measurements in TBI patients; monitoring duration > 6 h; glucose, lactate, and K | + | levels in spreading depolarisation (K+ was measured by an ion-selective electrode). | |||||||||||
Gowers et al. (2019) [52] | Enzymatic-electrochemical | In-vivo (human) | Detected a sudden surge of lactate levels during continuous online dialysate measurements in TBI patients. | ||||||||||
Gifford et al. (2021) [51] | Enzymatic-electrochemical | In-vivo (human) | Reported declining glucose levels in 3 TBI patients, and persistent low glucose in 1 TBI patient, in dexamethasone-enhanced continuous online microdialysis. | ||||||||||
Alimagham et al. (2021) [49] | Optical | (mid-IR) | Ex-vivo (human) | Microdialysate measurements from TBI patients, offline. LoDs of 0.5, 0.2, and 0.1 mM for glucose, lactate, and pyruvate respectively. Quantification of brain metabolites was compared with a conventional enzymatic-colorimetric microdialysis analyser (ISCUSflex). |