Astrocytes are essential players of brain circuit development and homeostasis controlling many aspects of synapse formation, function, plasticity and elimination both during development and adulthood. Accordingly, alterations in astrocyte morphogenesis and physiology may severely affect proper brain development, causing neurological or neuropsychiatric conditions. Recent findings revealed a huge astrocyte heterogeneity among different brain areas, which is likely at the basis of the different synaptogenic potential of these cells in selected brain regions.
Astrocytes are essential players of brain circuit development and homeostasis controlling many aspects of synapse formation, function, plasticity and elimination both during development and adulthood. Accordingly, alterations in astrocyte morphogenesis and physiology may severely affect proper brain development, causing neurological or neuropsychiatric conditions. Recent findings revealed a huge astrocyte heterogeneity among different brain areas, which is likely at the basis of the different synaptogenic potential of these cells in selected brain regions. This review highlights recent findings on novel mechanisms that regulate astrocyte-mediated synaptogenesis during development and the control of synapse number in the critical period or upon synaptic plasticity.
- astrocyte factors
- astrocyte diversity
- synaptogenesis
- synaptic pruning
- synaptic plasticity
1.IControducention
. Introduction/overview.
Astrocytes constitute the most abundant glial cells in the human brain [1]. A single cortical astrocyte enwraps multiple neuronal cell bodies and dendrites. It contacts more than 100,000 synapses through its finer perisynaptic astroglial processes (PAPs) [2, 3].
Through these contacts, astrocytes monitor and modulate synaptic function, thus actively controlling synaptic transmission. The close structural and functional interaction of the perisynaptic astrocytic processes with the neuronal pre- and postsynaptic structures led to the “tripartite synapse” concept [4] with astrocytes modulating synaptic transmission via the release of gliotransmitters, such as glutamate, D-serine, and ATP, and neurotransmitter reuptake from the active zone. The “tripartite synapse” concept introduced to the scientific community the key role of astrocytes in information transfer and storage in the adult brain (see reviews [5-8]).
However, the relation between astrocytes and neurons starts already during brain development, when astrocytes play key roles in neuronal circuit formation, in particular controlling synapse assembly and maturation, as well as synaptic refinement. In this review, we will provide an updated summary of the astrocyte-secreted molecules involved in synaptogenesis, and we will focus on the recently discovered mechanisms that regulate astrocyte-mediated synaptogenesis. We will also present an overview of how astrocytes exert a tight control over synapse number, which goes well beyond the synaptogenic period, affecting structure and function of the mature synapse.
Accumulating evidence indicate that alterations astrocyte morphogenesis and physiology in this period may severely affect proper brain development, causing neurological or neuropsychiatric conditions. In particular by using patients-derived astrocytes, recent studies unveiled the potential involvement of these cells in the pathophysiology of neurodevelopmental disorders, such as Rett and Down syndromes. Neurons cultured with astrocytes differentiated from Rett syndrome patients-specific iPSCs shows synaptic defects [9] whereas astrocytes differentiated from Down syndrome patients-specific iPSCs display altered transcriptomic profile, altered adhesion capability and increased cellular dynamics, and may impact on neuronal synaptogenesis [10, 11]. Not only genetic defects but also lengthy use of general anaesthetics in infants has been reported to affect astrocyte morphogenesis and associate to neurobehavioral deficits [12]. In this review, we also reported those evidence that link specific astrocyte factors, involved in synaptogenesis or in the regulation of synaptic function, to neurological and neuropsychiatric conditions.
- Astrocyte-secreted factors induce synaptogenesis.
The synaptogenic role of astrocytes was initially discovered using a purified retinal ganglion cell (RGC) culture system. RGC neurons grown in the absence of astroglia form very few synapses. However, synapse formation is increased upon addition of astrocyte-conditioned media (ACM) [13]. Since then, a large number of studies demonstrated that astrocytes are essential players in promoting synaptogenesis, particularly during brain development, and provided evidence for many astrocyte-secreted factors, including proteins, lipids, and small molecules that control different aspects of excitatory and inhibitory synapse formation and maturation (Table 1)
Table 1. Main synaptogenetic astrocyte-secreted molecules.
|
|
Name |
Type of synapse |
Molecular pathway |
Effects on synapses |
References |
|
Synapse formation |
|
|
|
|
|
|
|
TSP 1&2 |
excitatory |
α2δ-1-> Rac |
Promote silent synapses formation and actin remodelling at the spine |
[14-16] |
|
|
Hevin |
excitatory |
presynaptic NRX1a and postsynaptic NL1B |
Promotes silent synapses formation |
[17-19] |
|
|
SPARC |
excitatory |
dominant-negative on hevin neuronal partners |
Antagonizes hevin-induced synapses |
[17, 20] |
|
|
ApoE/cholesterol |
excitatory |
steroid and hedgehog pathways |
Promotes excitatory synapses formation and increases presynaptic strength and release probability |
[21, 22] |
|
|
BDNF |
excitatory |
erbB |
Promotes excitatory synapses formation |
[23] |
|
|
Estrogen |
excitatory |
ER-α receptor |
Promotes excitatory synapses formation |
[24] |
|
|
γ-protocadherin |
excitatory & inhibitory |
astrocytes/neurons contact |
Promotes excitatory synapses formation |
[25] |
|
|
TGFβ |
excitatory & inhibitory |
NMDAR/serine D/CAMKII |
Promotes excitatory and inhibitory synapses formation |
[26-28] |
|
Synapse maturation |
|
|
|
|
|
|
|
PTX3 |
excitatory |
β3 integrin/MAPK |
Leads to functional activation of GluA-containing silent synapses |
[29] |
|
|
Glypican 4&6 |
excitatory |
RPTPδ/NP1/GluA1 recruitment |
Induce functional synapses |
[30, 31] |
|
|
Chordin-like 1 |
excitatory |
through CR repeats, but BMP indipendent |
Induces maturation in GluA2-containing synapses |
[32] |
|
|
Glial neuroligins (1&2) |
excitatory & inhibitory |
neuronal neurexins |
Promote AMPA and NMDA receptors recruitment |
[33, 34] |
|
|
Wnt |
NMJ |
Repo (Drosophila) |
Increases synaptic AMPAR |
[35] |
|
Other |
|
|
|
|
|
|
|
Ephrin A3 |
excitatory |
Rac |
Promotes normal dendritic spine morphology |
[36, 37] |
|
|
Semaphorin 3A |
excitatory & inhibitory |
plexin A/neuropilin 1 receptor complexes |
Positional cue required for proper establishment of motor neuron and sensory neuron circuit formation |
[38] |
|
|
Neuregulin 1 |
excitatory & inhibitory |
erbB |
Guides tangential migration of cortical GABAergic interneurons and radial migration of differentiating pyramidal neurons |
[39] |
|
|
BMP |
excitatory & inhibitory |
BMP receptor |
Maintains the homeostasis of the synaptic microenvironment |
[40] |
|
|
Maverick |
NMJ |
Gbb-dependent retrograde signaling (Drosophila) |
Coordinates pre- and postsynaptic maturation |
[41] |
The synaptogenic role of astrocytes was initially discovered using a purified retinal ganglion cell (RGC) culture system. RGC neurons grown in the absence of astroglia form very few synapses. However, synapse formation is increased upon addition of astrocyte-conditioned media (ACM)[1]. Since then, a large number of studies demonstrated that astrocytes are essential players in promoting synaptogenesis, particularly during brain development, and provided evidence for many astrocyte-secreted factors, including proteins, lipids, and small molecules that control different aspects of excitatory and inhibitory synapse formation and maturation. During the last two decades the astrocyte
During the last two decades the astrocyte
secretome, i.e. entire set of secreted proteins according to the definition coined by Tjalsma et al., in 2000[2][3], has been extensively analyzed in search of synapse-promoting factors. The full list of astrocytes’ molecules involved in synaptogenesis is reported in Table 1 and has been extensively reviewed in the last years, (see also [4][5][6][7]). Here we focus on three classes of well-known synaptogenic factors, which play key roles in early synaptogenesis through recently discovered molecular mechanisms.
, i.e. entire set of secreted proteins according to the definition coined by Tjalsma et al., in 2000 [42, 43], has been extensively analyzed in search of synapse-promoting factors. The full list of astrocytes’ molecules involved in synaptogenesis is reported in Table 1 and has been extensively reviewed in the last years, (see also [44-47]). Here we focus on three classes of well-known synaptogenic factors, which play key roles in early synaptogenesis through recently discovered molecular mechanisms.
2. Thrombospondins and Pentraxin 3.
2.1. Thrombospondins and Pentraxin 3.
In the first two weeks of postnatal mouse development, which correspond to a period of massive synaptogenesis in most brain areas, astrocyte-derived thrombospondin 1 and 2 (TSP1, TSP2) are highly expressed and initiate formation of synapses[8]. This process occurs through TSP binding membrane proteins, including the Ca
In the first two weeks of postnatal mouse development, which correspond to a period of massive synaptogenesis in most brain areas, astrocyte-derived thrombospondin 1 and 2 (TSP1, TSP2) are highly expressed and initiate formation of synapses [14]. This process occurs through TSP binding membrane proteins, including the Ca
2+ channel subunit α2δ-1[9] or the postsynaptic cell adhesion molecule Neuroligin1[10]. It has been demonstrated that TSPs induce the formation of ultrastructurally normal synaptic contacts, which are presynaptically active with cycling synaptic vesicles. Remarkably, they result postsynaptically silent because of the lack of functional 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA) receptors[8].
channel subunit α2δ-1 [15] or the postsynaptic cell adhesion molecule Neuroligin1 [48]. It has been demonstrated that TSPs induce the formation of ultrastructurally normal synaptic contacts, which are presynaptically active with cycling synaptic vesicles. Remarkably, they result postsynaptically silent because of the lack of functional 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA) receptors [14].
An elegant study by Risher and colleagues recently demonstrated that in the cerebral cortex TSP–α2δ-1 interaction controls synaptogenesis by acting postsynaptically via Rac1[11]. Indeed, they found that the synaptogenic function of α2δ-1 is cell autonomous to neurons, requiring the postsynaptic expression of α2δ-1. Upon TSP–α2δ1 interaction at the postsinaptic level, the guanine nucleotide exchange factors (GEFs), Kalirin-7 and β-Pix/Cool-1 β, activate Rac1 pathway and promote the remodeling of actin cytoskeleton at the nascent synaptic contact[11]. TSP1 and TSP2 display a developmentally regulated pattern of expression and are downregulated around the second and third weeks of postnatal development[8].It has been known for a long time that the activity of TSPs is fundamental to promote the formation of early synapses, which are silent. Which is/are the factors involved in the activation of these first synapses was not known until very recently. Indeed, it has been recently demonstrated that concomitantly to TSP secretion, astrocytes release the innate immune molecule pentraxin3 (PTX3), which promotes the functional maturation of excitatory synapses formed during the first wave of synaptogenesis by inducing AMPA receptors clustering at the synapse[12].
An elegant study by Risher and colleagues recently demonstrated that in the cerebral cortex TSP–α2δ-1 interaction controls synaptogenesis by acting postsynaptically via Rac1 [16]. Indeed, they found that the synaptogenic function of α2δ-1 is cell autonomous to neurons, requiring the postsynaptic expression of α2δ-1. Upon TSP–α2δ1 interaction at the postsinaptic level, the guanine nucleotide exchange factors (GEFs), Kalirin-7 and β-Pix/Cool-1 β, activate Rac1 pathway and promote the remodeling of actin cytoskeleton at the nascent synaptic contact [16]. TSP1 and TSP2 display a developmentally regulated pattern of expression and are downregulated around the second and third weeks of postnatal development [14]. Concomitantly, glial secretion of TSP4 increases [49]. TSP4 differs from TSP1 and TSP2 for the lack of the procollagen and properdin-like (type I) repeats that enable interactions with glycosaminoglycans, integrin-associated proteins and promote the activation of TGFβ [50]. Furthermore, whereas TSP1-deficient and TSP2-deficient mice display a significant decrease in the frequency of excitatory synapses [14, 51], TSP4-deficient mice show increased synaptic activity due to the lack of regulation of presynaptic Ca2+ channels [52]. Thus, the developmental shift of TSPs subtypes’ activity during brain development may affect different aspects of synaptogenesis that need to be further explored. It has been known for a long time that the activity of TSPs is fundamental to promote the formation of early synapses, which are silent. Which is/are the factors involved in the activation of these first synapses was not known until very recently. Indeed, it has been recently demonstrated that concomitantly to TSP secretion, astrocytes release the innate immune molecule pentraxin3 (PTX3), which promotes the functional maturation of excitatory synapses formed during the first wave of synaptogenesis by inducing AMPA receptors clustering at the synapse [29].
This occurs through a process involving the key PTX3 binding partner, TSG6, the remodeling of the perineural extracellular matrix surrounding synaptic contacts and requires β1-integrins. Quite unexpectedly, PTX3 deficient mice display weaker excitatory synapses in the hippocampus not only at young ages but also at P30, indicating that lack of endogenous PTX3 results in synapse defects, which apparently cannot be rescued by other astrocyte-derived factors expressed at later developmental stages[13][14][15], see next paragraphs. Notably, PTX3 is able to interact with TSP1 and 2, but not with TSP4. Upon binding to TSP1, PTX3 activity is inhibited, thus representing an additional mechanism of control of the process[12]. Notably, PTX3 and TSP1 display a spatially and temporally overlapped expression also in human brain, being higher in the astrocytes of foetal cerebral cortex[16]. The relative amount of the two molecules could therefore be crucial to set the proper balance between synaptic growth and synapse maturation during the period of early synaptogenesis. Both common and rare variants of the gene encoding for TSP1,
This occurs through a process involving the key PTX3 binding partner, TSG6, the remodeling of the perineural extracellular matrix surrounding synaptic contacts and requires β1-integrins. Quite unexpectedly, PTX3 deficient mice display weaker excitatory synapses in the hippocampus not only at young ages but also at P30, indicating that lack of endogenous PTX3 results in synapse defects, which apparently cannot be rescued by other astrocyte-derived factors expressed at later developmental stages [17, 30, 31], see next paragraphs. Notably, PTX3 is able to interact with TSP1 and 2, but not with TSP4. Upon binding to TSP1, PTX3 activity is inhibited, thus representing an additional mechanism of control of the process [29]. Notably, PTX3 and TSP1 display a spatially and temporally overlapped expression also in human brain, being higher in the astrocytes of foetal cerebral cortex [53]. The relative amount of the two molecules could therefore be crucial to set the proper balance between synaptic growth and synapse maturation during the period of early synaptogenesis. Both common and rare variants of the gene encoding for TSP1,
THBS1, have been found to be associated with Autism Spectrum Disorders (ASD) in a cohort of 313 patients by Sanger sequencing[17]. Furthermore single nucleotide polymorphisms (SNPs) of PTX3 gene have been reported to result in PTX3 deficiency in humans and are associated with changed susceptibility to infections and altered inflammatory response[18][19][20]. Whether these PTX3 variants might be linked to altered synaptogenesis under physiological conditions or in response to an inflammatory insult occurring during brain development is presently not known.
, have been found to be associated with Autism Spectrum Disorders (ASD) in a cohort of 313 patients by Sanger sequencing [54]. Furthermore single nucleotide polymorphisms (SNPs) of PTX3 gene have been reported to result in PTX3 deficiency in humans and are associated with changed susceptibility to infections and altered inflammatory response [55-57]. Whether these PTX3 variants might be linked to altered synaptogenesis under physiological conditions or in response to an inflammatory insult occurring during brain development is presently not known.
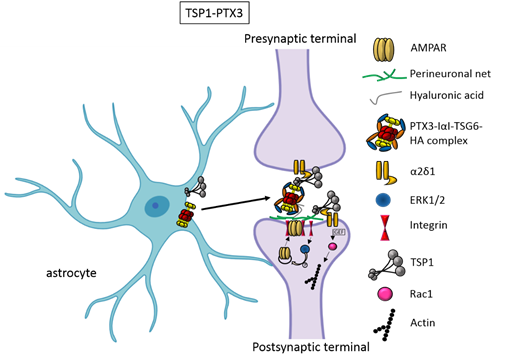
Figure 1. TSP1 TSP1 and PTX3 cooperate to promote early formation of functional excitatory synapses. TSP1 binds to the α2δ1 receptor on the presynaptic terminals stimulating an increase of structurally normal but silent synapses and to the α2δ1 receptor at the postsynaptic site activating Rac1 and stimulating actin remodelling to promote spinogenesis. PTX3 promotes the functional maturation of these synapses by recruiting AMPA receptors at the synapse.
and PTX3 cooperate to promote early formation of functional excitatory synapses. TSP1 binds to the α2δ1 receptor on the presynaptic terminals stimulating an increase of structurally normal but silent synapses and to the α2δ1 receptor at the postsynaptic site activating Rac1 and stimulating actin remodelling to promote spinogenesis. PTX3 promotes the functional maturation of these synapses by recruiting AMPA receptors at the synapse.
In addition to PTX3, another glial factor which increases AMPA receptors (AMPAR) levels at synapses has been recently identified in the astrocyte secretome, Chordin-like 1 (Chrdl1), whose expression in the cerebral cortex peaks at P12–P14, a bit later than PTX3. In particular, Chrdl1 has been shown to stimulate the insertion of GluA2-containing calcium impermeable AMPARs at the synapse thus regulating the so-called AMPAR switch underlying the functional maturation of excitatory synapses in the cerebral cortex [32].
3. Hevin and SPARC.
2.2. Hevin and SPARC.
Besides TSPs, two members of the secreted protein acidic, enriched in cysteine (SPARC) family proteins, Hevin (also known as SPARC-like1/SPARCL1) and SPARC were recognised as astrocyte-secreted factors that control synapse formation between cultured RGCs. Similar to TSPs, treatment of RGCs with purified Hevin is sufficient to induce formation of structurally mature, but postsynaptically silent synapses[15]. Analyses of developing visual cortices in Hevin-null mice revealed fewer thalamocortical synapses and spines displaying features of immaturity, indicating that the protein is required for proper development of the thalamocortical synaptic connectivity[21]. Astrocyte-derived Hevin has been demonstrated to serve as a bridge for the non-interacting isoforms of neurexin-1alpha and neuroligin-1B at the level of thalamocortical connections in the developing visual cortex[22]. By contrast, SPARC antagonizes the synaptogenic action of Hevin through a dominant-negative action, most likely by interfering with the ability of Hevin to bridge neurexin-1alpha and neuroligin-1B[15]. In addition, SPARC is able to trigger a cell-autonomous programme of synapse elimination[23]. In the developing mouse brain, Hevin and SPARC are expressed at high levels in astrocytes during the second and third weeks, which coincide with periods of synapse stabilization and synaptic refinement. Of note,
Besides TSPs, two members of the secreted protein acidic, enriched in cysteine (SPARC) family proteins, Hevin (also known as SPARC-like1/SPARCL1) and SPARC were recognised as astrocyte-secreted factors that control synapse formation between cultured RGCs. Similar to TSPs, treatment of RGCs with purified Hevin is sufficient to induce formation of structurally mature, but postsynaptically silent synapses [17]. Analyses of developing visual cortices in Hevin-null mice revealed fewer thalamocortical synapses and spines displaying features of immaturity, indicating that the protein is required for proper development of the thalamocortical synaptic connectivity [18]. Astrocyte-derived Hevin has been demonstrated to serve as a bridge for the non-interacting isoforms of neurexin-1alpha and neuroligin-1B at the level of thalamocortical connections in the developing visual cortex [19]. By contrast, SPARC antagonizes the synaptogenic action of Hevin through a dominant-negative action, most likely by interfering with the ability of Hevin to bridge neurexin-1alpha and neuroligin-1B [17]. In addition, SPARC is able to trigger a cell-autonomous programme of synapse elimination [58]. In the developing mouse brain, Hevin and SPARC are expressed at high levels in astrocytes during the second and third weeks, which coincide with periods of synapse stabilization and synaptic refinement. In the adult brain, SPARC is downregulated whereas Hevin expression remains high [17], in line with Hevin role also in synaptic maintaninance and repair [59] as well as in synaptic plasticity (see below, chapter “Astrocyte-derived molecules affect synaptic plasticity”). Of note,
SPARCL1 has been recently identified as part of a network of genes linked to neuronal damage in the preclinical stages of Alzheimer’s disease (AD)[25]. In particular,
has been recently identified as part of a network of genes linked to neuronal damage in the preclinical stages of Alzheimer’s disease (AD) [60]. In particular,
SPARCL1 variants that correlate with lower gene expression levels in brain are associated with accelerated cognitive decline during preclinical AD.
variants that correlate with lower gene expression levels in brain are associated with accelerated cognitive decline during preclinical AD.
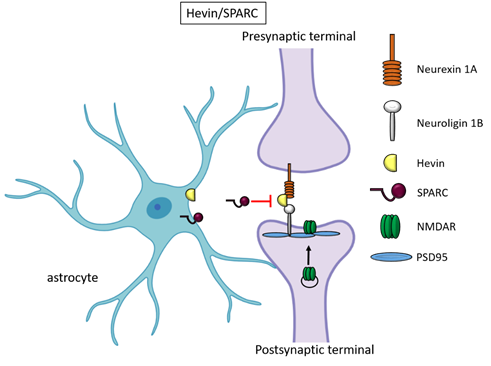
Figure 2. Hevin and SPARC. Astrocyte-secreted Hevin bridges presynand SPARCptic neurexin and postsynaptic neuroligin to favour the recruitment of PSD95 and NMDAR subunits at the synapse. SPARC on the contrary antagonizes Hevin effect with a still unknown mechanism.
2.3. Glypicans and neuronal pentraxins.
Recently, astrocyte-secreted glypicans 4 and 6 have been discovered by using the same experimental setting (i.e. RGC cultures and ACM) that led to the distrocytecovery of thrombospondins. Glypicans are expressed at later stages of development (second and third postnatal weeks). Exposure of RGC cultures to glypicans 4 or 6 resulted in enhanced formation of active excitatory synapses containing GluA1 receptors at difference to the action of TSPs [30]. GPC4 deficient mice show defective excitatory synapses in the hippocampus [30] and alteration in social behaviour which resemble ASD core symptoms and that can be linked with loss of GluA1 [61]. The ability of glypican 4 to induce active synapses involves neuronal pentraxin 1 (NP1) which is released from presynaptic terminals by signaling through presynaptic protein tyrosine phosphatase receptor delta (PTPRδ). NP1 then stimulates AMPA receptors clustering on the postsynaptic dendrite thus allowing the assembly of functional synapses [31].
Neuronal pentraxins - the membrane-bound "neuronal pentraxin receptor" (NPR) and the secreted proteins NP1 and NARP (i.e., NP2) - are expressed by neurons and promote AMPA clustering at excitatory synapses by direct binding of the N- terminal domain of the receptor [62-68]. Recently it has been demonstrated that knockdown of NPR in hippocampal neurons dramatically decreases assembly and function of both excitatory and inhibitory postsynaptic specializations [69]. Evidence showed that NPR recruits and stabilizes NP1 and NARP on the presynaptic plasma membrane suggesting that neuronal pentraxins act as trans-synaptic organizers of both excitatory and inhibitory synapses. Diseases associated with GPC4 include Simpson-Golabi-Behmel Syndrome which in some cases results in mild to severe intellectual disability [70].
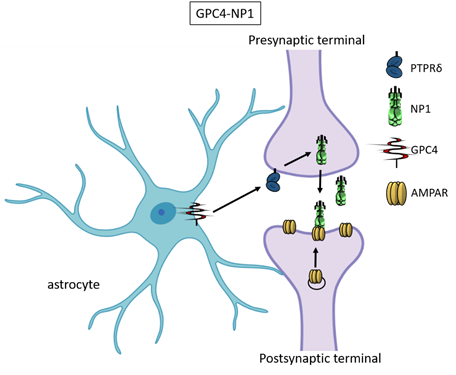
Figure 3. Glypican 4 and Neuronal Pentraxin 1. Astrocyte-derived Glypican 4 induces NP1 release from neurons through the PTPRδ receptor. NP1 stimulates AMPA receptors clustering on the postsynaptic terminal making the synapse functional.
- Astrocyte diversity affects their own synaptogenic potential.
Accumulating evidence indicate that the synaptogenic properties of astrocytes from diverse brain regions is different, indicating the specialization of local astrocyte populations within specific synaptic circuits. In line with specific astrocyte secretome varying according to the brain region, it has been reported that astrocytes from the cerebellum induce more synapses with respect to astrocytes from the midbrain, hippocampus or cortex, due to the higher expression of Hevin and glypican 4 in this population [71].
While astrocyte diversity has remained for a long time poorly investigated because of technical issues hamperidges presynaptic neurexin and postsyng their isolation from specific brain regions, recently developed techniques for single cell RNA profiling and proteomic studies largely advanced this line of investigation [49, 72, 73]. For instance, the fusion of reporter tags to ribosomal subunits allowed the isolation and analysis of actively translated mRNAs from astrocytes [72, 74, 75] while the use of magnetic-activated cell sorting preserved astrocyte morphology better than FACS [76, 77]. Taking advantage of this technical improvement, Batiuk and colleagues recently identified, in the adult brain, molecularly and functionally diverse subpopulations of astrocytes, which eventually exert different roles in supporting synaptogenesis [78]. In particular they recognised, throughout the cortex, astrocyte subtypes 2 and 3 (AST2 and 3), which differentially express genes involved in neurotransmission. AST2 is enriched in transcripts linked to glutamatergic neurotransmission, while AST3 is enriched in transcripts associated with GABAergic neurotransmission, thus suggesting that neuronal-derived signals induce astrocyte diversity at the local level [78].
Although deciphering the mechaptic neuroligin to favour the recruitment ofnisms responsible for the acquisition of astrocyte molecular and functional diversity is at its infancy, with intrinsic astrocytic factors and extrinsic neuronal signals being likely involved [79], it is becoming evident that the astrocyte specialization -in space and time- dynamically regulates the synaptogenic potential of these cells. This mainly occur through modifications of the astrocyte secretome composition throughout the lifespan. Of note, the synaptogenic effect of astrocytes was found to rely on the prevalence of a specific astrocyte population subtype, called population ‘C’, that is characterized by the presence of surface markers CD51, CD63 and CD71 [80] . This astrocyte subpopulation, isolated from ALDH1L1–eGFP adult mouse model, is highly enriched for genes associated with synapse assembly and function. Consistently, when co-cultured with cortical neurons, C astrocytes are able to increase the frequency of both cortical excitatory and inhibitory currents [80].
- Astrocytes control synaptic pruning.
Neurons generate an excess of synapses during development, some of them are subsequently eliminated to reach a precise neuronal circuit assembly through a process called “pruning” [46, 81]. This synaptic refinement happens in a precise time window defined “developmental critical period”, a brief interval where neural circuits can be modified by sensory inputs. Within critical periods, the unnecessary and weak synapses are eliminated, whereas the remaining inputs are further strengthened to form the mature neural circuit. Afterwards, synapse elimination continues in the mature nervous system, through experience-dependent structural synaptic plasticity, even if the number of elimination events drops with age [82].
Traditionally, microglia have been thought as the only glial cells responsible for synaptic pruning [83], but accumulating evidence showed that also astrocytes exert a key role in regulating synaptic elimination during development [34, 45, 84]. The phagocytic ability of astrocytes has been demonstrated previously both in vitro, by using cell cultures, and in vivo in models of injury [44]. For example, in glaucoma, astrocytes have been demonstrated to constitutively perform phagocytosis through internalizations of large portions of axonal cytoplasm and axon-derived organelles [85]. Interestingly, similarly to microglia [86, 87], the phagocytic activity of astrocytes appears to be influenced by the sex, as recently demonstrated by the use of synthetic steroids which induce a significantly higher phagocytic activity in astrocytes derived from female mice under resting and inflammatory conditions [88].
The astrocyte-mediated phagocytosis of synapses, which is driven by network activity and is crucial in shaping the circuit in response to experience, may involve direct or indirect mechanisms. Direct phagocytosis of supernumerary synapses in the developing brain occurs via the astrocyte phagocytic receptors MERTK and MEGF10 [89]. Both these pathways are activated by an “eat-me” signal, like phosphatidylserine exposure, as it occurs in case of microglia [90]. MEGF10 (Multiple EGFLike Domains 10) is a known mediator of phagocytosis. Its action involves different intracellular proteins but the exact function of this protein is still unknown [82]. MERTK (Mer proto-oncogene Tyrosine Kinase) exploits the integrin pathway to control the actin cytoskeleton during phagocytosis. These pathways have been found to be active in the retinogeniculate system, and indeed MEGF10 and MERTK deficient mice display defective synaptic pruning and, therefore, impaired eye specific segregation in the dorsolateral geniculate nucleus (dLGN) [46, 82, 89].
Astrocytes can also promote indirect phagocytosis by mediating the neuronal expression of phagocytic markers recognized by microglia. With this respect, the pioneer study by Bialas and Stevens showed that astrocytes indirectly regulate synapse elimination via the secretion of the transforming growth factor-β (TGF-β) in the retina. Astrocytes release TGFβ that stimulates C1q expression in RGC neurons. C1q is the initiating protein of the classical complement cascade that starts the complement-dependent synaptic refinement in the retinogeniculate system mediated by microglia. Accordingly, inhibition of TGFβ resulted in defects in eye-specific segregation in the dLGN resembling microglia-deficient phenotypes [91, 92].
In the last years direct evidence linking synapse elimination with human diseases has been emerging. Using reprogrammed in vitro model of microglia-mediated synapse engulfment, increased synapse elimination in patient-derived neural cultures and isolated synaptosomes was demonstrated [93]. This evidence added to more indirect indications supporting a link between defective synapse refinement and human diseases. Indeed, altered synaptic pruning has been reported in mice models with mutations of PTEN, which accounts for ~10% of cases of ASD95 [94]. It has been demonstrated that the genetic lack of the microglia receptor TREM2 impairs the ability of microglia to eliminate synapses and results in ASD-like phenotype and that an impairment in synaptic material degradation in atg7-deficient mice microglia causes social behavioural defects and repetitive behaviours typical of ASDs [95].
Consistently, alterationd s in synapse number and structure and abnormalities in glial cells functioning have been found in many neurological disorders [96-100]. Given the central role of astrocytes, together with microglia, in synapse elimination and based on the tight cooperation between astrocytes and neurons in brain development and function, it is reasonable to hypothesize that alterations in the astrocyte-dependent synaptic pruning could be a concurring factor in brain pathologies. Despite the difficulty in studying human pathologies and the paucity of tools to investigate specifically astrocytes hampered this line of investigation in the past, important contributions to this field are expected in the future.
Another new evidence for indirect mechanism of phagocytosis involves the proinflammatory cytokine Interleukin33 (IL33), which is expressed specifically by astrocytes in the CNS during the critical period of synaptic refinement. Vainchtein and colleagues demonstrated that IL33 directly mediates the developmentally regulated removal of excitatory connections between the reticular and the ventrobasal nuclei of the thalamus, increasing microglial phagocytic ability. Indeed, astrocyte-IL33 expression increases following synaptic maturation, suggesting a homeostatic loop by which astrocytes react to increased synapse number promoting the process of synaptic pruning operated by microglia. On the contrary, IL33 deficiency has been associated with an excessive number of excitatory synapses and overall hyperexcitable intrathalamic circuit compared to wild-type controls [101-103]. The mechanism by which astrocyte-derived IL33 regulates microglial synaptic function is a new function for this cytokine best known as an “alarmin” involved in tissue homeostasis. It is released by dying cells and is crucially involved in wound healing and in the inflammatory response after injury [38]. IL33 retains a role of “alarmin” also in the adult CNS, in fact in a mouse model of spinal cord injury IL33 is upregulated by spinal cord astrocytes and promotes the resolution of inflammation through a mechanism involving TNFα [104].
- Astrocyte-derived molecules affect synaptic plasticity.
5.1. Role of astrocytes in critical period.
Some glial factors with a recognized synaptogenic role at early stages of development are involved also in the subsequent phase of synapse refinement during critical period. For example, Hevin expression remains high during the critical period [17] and it is required for the proper establishment of thalamocortical synapses. Hevin-deficient mice display an increased number of immature spines and altered localization of excitatory synapses indicating that lack of Hevin results in abnormal thalamocortical connectivity probably due to the defective synaptic refinement process [18]. Also, Hevin acts as a bridge between the neuronal neurexin 1α and neuroligin 1B which do not interact directly. This interaction recruits NMDA receptors at the synapse. Mice lacking Hevin display impaired ocular dominance plasticity, an event where synapses in the visual cortex remodel in response to changed visual experience. R subunits at the synapse. escue of Hevin expression specifically in astrocytes of the visual cortex before the closure of critical period restores ocular dominance plasticity, showing that expression of Hevin in astrocytes is sufficient to control this form of plasticity [19]. Furthermore, neuroligins are also expressed by astrocytes and through their neuronal partner neurexin 1 control astrocytic morphogenesis and excitatory synaptogenesis [34]. A recent finding proposes astrocytic neuroligins to ensure proper critical period termination in the developing Drosophila motor circuit. Indeed in mutant drosophila, reduced neuroligin-neurexin signalling results in dendritic microtubules destabilization, enhanced dendrite dynamics, and impaired locomotor behaviour [105].
Chordin-like 1 (Chrdl1) is another example of astrocyte secreted protein which is expressed throughout postnatal development and in the critical period. Besides being involved in the maturation of excitatory AMPAR-containing synapses during synaptogenesis it plays a key role in regulating the closure of critical period in the maturing brain. In the absence of Chrdl1, synapses in the visual cortex do not undergo the physiological switch from calcium-permeable to GluA2-containing calcium-impermeable channels and therefore they contain less GluA2 [106-108]. As a consequence, the removal of one eye after the closure of the critical period (monocular enucleation) in adult Chrdl1 deficient mice resulted in a significant increased plasticity and remodelling in the binocular zone of mutant mice with respect to wild type. The loss of Chrdl1 results therefore in an extended critical period plasticity that remains open up to adulthood [109]. In humans, many different mutations in Chrdl1 that cause X-linked megalocornea1, a disease characterized by enlarged corneas and an increased risk of presenile cataracts, have been identified. Unexpectedly, these patients display a superior verbal IQ, verbal memory, and executive skills [110].
5.2. Role of astrocytes in adult forms of synaptic plasticity.
Synaptic plasticity is the ability of a synapse to modify its structure in response to external stimuli, which persists through all the life and allow to continuously modifying brain circuits in response to experience. Synapses can increase their strength (long term potentiation, LTP) or decrease their strength (long term depression, LTD) in response to alterations in neuronal activity. Synaptic scaling is a form of homeostatic plasticity with compensatory up-regulation of synaptic activity in response to prolonged periods of activity deprivation. Both forms of plasticity play together in a concerted fashion and astrocytes recently came out as possible candidate to orchestrate these forms of plasticity, for further insights on this topic see [111].
In neurons, one of the major structural change following LTP or LTD concerns the appearance or retraction of dendritic spines and therefore the change of synapse number upon LTP or LTD induction. Similarly, astrocytic processes undergo rapid changes in volume and motility. Astrocyte PAPs are extremely sensitive to activity-driven changes at the synapse. Induction of LTP transiently enhances the motility and retraction of PAPs allowing growth of the postsynaptic dendritic spine to occur. Subsequently, they will adjust their coverage to adapt to the new structure of the synapse [112, 113].
The glial factor SPARC, on the contrary analready mentioned for its role in controlling synaptogenesis, is also fundamental to limit the number of synaptic AMPAR levels in the adult. SPARC KO mice in fact display an abnormal accumulation of surface AMPAR at the synapse and enhanced excitatory glutamatergic neurotransmission, resulting in an altered NMDAR/AMPAR synaptic ratio and impaired LTP [20].
In addition, ephrinB1, which is expressed in agonizes dult mice, acts as a regulator of synaptogenesis in adult hippocampus and affects mouse learning behaviours. Of note, ephrinB1-deficient mice display immature dendritic spines, reduced evoked synaptic firing in CA1 hippocampal neurons and proper acquisition of fear memory, but enhanced contextual fear memory recall. Moreover, astrocytic ephrin-B1 competes with the neuronal ephrin-B1 and triggers astrocyte-mediated engulfment of ephrin B receptor-containing synapses via trans-endocytosis. This study demonstrated that astrocyte ephrinB1 is crucial to limit new synapse formation in the adult hippocampus upon learning processes [114]. The same group, using an ephrin1B overexpressing mouse model, recently investigated the mechanism through which astrocytic ephrin 1B influences learning process. They demonstrated that alterations in contextual fear learning processes are characterized by reduced formation of new spines more than impaired spine maturation [115]. Furthermore, astrocyte-derived ephrin 1B regulates synapse remodelling in the hippocampus in a mouse model of traumatic brain injury through an increased expression of STAT3 in astrocytes [116].
Finally, new interesting roles in synaptic plasticity are emerging for lipid components that are produced and released by astrocytes, including astrocyte-secreted cholesterol. Astrocytic cholesterol synthesis is the primary source of cholesterol in the CNS. Its function is not confined to support synaptogenesis [117], but it has a fundamental role also in synaptic plasticity. The regulatory element binding proteins (SREBPs) control cholesterol production ensuring an appropriate level in the brain. In the hippocampus, reduced cholesterol synthesis affects spines development and modification. In fact, in mice defective for SREBPs both short term and long-term plasticity are impaired [46, 117].
Alterations in astrocyte lipid metabolism are associated with synaptic dysfunctions. For example, in Huntington diseasevin effect with a stil, mutant huntingtin protein in astrocytes decreases SREBP maturation leading to impaired cholesterol biosynthesis and secretion, thus affecting synapse number and activity [118]. Cholesterol metabolism is significantly altered in Huntington’s disease patients. These alterations are associated with a striking reduction of the cholesterol 24-hydrolase (CYP46A1) expression in the putamen of patients. CYP46A1 is crucial for brain cholesterol elimination, because it mediates cholesterol hydroxylation. Of note, a gene therapy approach with CYP46A1 has been recently tested in a mouse model of Huntington’s disease [119].
In addition, in the autosomal recessive Niemann-Pick disease type C (NPC), which is characterized by an accunmulation of unesterified cholesterol in late endosomes/lysosomes (LE/L), mutations in the NPC1 gene lead to impaired cholesterol transport in astrocytes. NPC affects neurological and psychiatric functions including learning difficulties and cognitive impairments, as well as various internal organs. Affected neurons display higher cholesterol content in the soma and reduced cholesterol content in the distal axons. It is conceivable that some of the neurological deficits in NPC disease might be due to a deficiency of cholesterol in axons [120].
Apolipoprotein E4 (apoE4) is the main carrier of cholesterol and it is the main genetic risk factor associated with sporadic Alzheimer’s disease (AD). It has a role inown mechanis controlling Aβ formation through regulation of lipid rafts function in vivo [121]. Indeed, cholesterol is found to be enriched in the brain plasma membranes of AD patients. The cholesterol level increases throughout the course of clinical disease, and more increase was observed when the disease progresses [53]. Because cholesterol is a multifunctional metabolite, abnormal cholesterol metabolism by ApoE4 would lead not only altered cholesterol transport to other cell types but also functional deficits in astrocytes as observed in neurons [122].
Regarding homeostatic plasticity, which allows neural circuits to elevate or reduce the activity of the full neuronal network to induce robust compensatory changes in the strength of excitatory and inhibitory synapses. It maintains appropriate levels of excitability and connectivity despite changes in the surrounding environment brought about by metabolism and experience-dependent plasticity. Without homeostatic synaptic scaling, neural networks can become unstable and perform suboptimally [123, 124]. Glial TNFα plays a fundamental role in this process by stimulating rapid exocytosis of AMPARs calcium-permeable GluA2-lacking AMPARs and the simultaneous endocytosis of GABA-A receptors thus strongly affecting the balance of excitation-to-inhibition (E/I balance) [44, 111, 125-128]. In addition to regulating post-synaptic receptors content glial TNF may also regulate pre-synaptic neurotransmitter release [125, 129, 130]. Consistently TNFα-deficient mice display normal LTP and LTD but they did not undergo homeostatic synaptic scaling demonstrating that TNFα is required for upregulation of AMPARs upon depression [131].
4. Glypicans and neuronal pentraxins.
- Conclusions.
Astrocytes are an essential “component” of the synapse, together with the pre- and postsynaptic compartments. They play important roles in synapse formation, maturation, and elimination, as well as in the regulation of many aspects of synaptic activity such as synaptic plasticity.
In this review, we presented an updated list of the astrocyte-secreted molecules fundamental for synaptogenesis. We focused on three main groups of molecules that act in concert to promote synaptogenesis, describing newly discovered mechanisms. Further, we went through some recent astrocyte-derived proteins and lipids implied in direct and indirect phagocytosis, which mediates synaptic refinement during the period of synaptic maturation. Last, we reviewed astrocytes’ contribution to the adult forms of synaptic plasticity.
The idea of “tripartite synapse” back in 1999 [132] proposed for the first time the contribution of astrocytes to synapse formation and function. Since then, many studies discovered a number of molecules involved in this process, but this increasing knowledge has still some gaps. For example, do these molecules act widespread into the brain or in region specific manner? Understanding in more detail the basis of astrocyte diversity would allow to thoroughly defining the specific role of each type. This would contribute to dissect also which functions are redundant and which cannot be compensated.
Moreover emerging evidence point to a role of astrocytes in human brain diseases, such as ASD and other neurodevelopmental diseases. This area of investigation is still in its infancy. In this respect, it will be important to investigate how astrocytes modulate synaptic development and function in the circuits that mediate cognition, emotion, and social function. At present, this is a very ambitious goal that could give unforeseen possibilities in the treatment of brain diseases.
Recently, astrocyte-secreted glypicans 4 and 6 have been discovered by using the same experimental setting (i.e. RGC cultures and ACM) that led to the discovery of thrombospondins. Glypicans are expressed at later stages of development (second and third postnatal weeks). Exposure of RGC cultures to glypicans 4 or 6 resulted in enhanced formation of active excitatory synapses containing GluA1 receptors at difference to the action of TSPs [13]. GPC4 deficient mice show defective excitatory synapses in the hippocampus[13] and alteration in social behaviour which resemble ASD core symptoms and that can be linked with loss of GluA1[26]. The ability of glypican 4 to induce active synapses involves neuronal pentraxin 1 (NP1) which is released from presynaptic terminals by signaling through presynaptic protein tyrosine phosphatase receptor delta (PTPRδ). NP1 then stimulates AMPA receptors clustering on the postsynaptic dendrite thus allowing the assembly of functional synapses[14].
Neuronal pentraxins - the membrane-bound "neuronal pentraxin receptor" (NPR) and the secreted proteins NP1 and NARP (i.e., NP2) - are expressed by neurons and promote AMPA clustering at excitatory synapses by direct binding of the N- terminal domain of the receptor[27][28][29][30][31][32][33]. Recently it has been demonstrated that knockdown of NPR in hippocampal neurons dramatically decreases assembly and function of both excitatory and inhibitory postsynaptic specializations[34]. Evidence showed that NPR recruits and stabilizes NP1 and NARP on the presynaptic plasma membrane suggesting that neuronal pentraxins act as trans-synaptic organizers of both excitatory and inhibitory synapses. Diseases associated with GPC4 include Simpson-Golabi-Behmel Syndrome which in some cases results in mild to severe intellectual disability[35].
References.
- Eroglu, C. and B.A. Barres, Regulation of synaptic connectivity by glia. Nature, 2010. 468(7321): p. 223-31.
- Halassa, M.M., T. Fellin, and P.G. Haydon, The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med, 2007. 13(2): p. 54-63.
- Heller, J.P. and D.A. Rusakov, Morphological plasticity of astroglia: Understanding synaptic microenvironment. Glia, 2015. 63(12): p. 2133-51.
- Araque, A., et al., Astrocyte-induced modulation of synaptic transmission. Can J Physiol Pharmacol, 1999. 77(9): p. 699-706.
- Weber, B. and L.F. Barros, The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb Perspect Biol, 2015. 7(12).
- Durkee, C.A. and A. Araque, Diversity and Specificity of Astrocyte-neuron Communication. Neuroscience, 2019. 396: p. 73-78.
- Hasan, U. and S.K. Singh, The Astrocyte-Neuron Interface: An Overview on Molecular and Cellular Dynamics Controlling Formation and Maintenance of the Tripartite Synapse. Methods Mol Biol, 2019. 1938: p. 3-18.
- Farhy-Tselnicker, I. and N.J. Allen, Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev, 2018. 13(1): p. 7.
- Williams, E.C., et al., Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum Mol Genet, 2014. 23(11): p. 2968-80.
- Ponroy Bally, B., et al., Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum Mol Genet, 2020. 29(5): p. 785-802.
- Araujo, B.H.S., et al., Down Syndrome iPSC-Derived Astrocytes Impair Neuronal Synaptogenesis and the mTOR Pathway In Vitro. Mol Neurobiol, 2018. 55(7): p. 5962-5975.
- Zhou, B., et al., Astroglial dysfunctions drive aberrant synaptogenesis and social behavioral deficits in mice with neonatal exposure to lengthy general anesthesia. PLoS Biol, 2019. 17(8): p. e3000086.
- Ullian, E.M., et al., Control of synapse number by glia. Science, 2001. 291(5504): p. 657-61.
- Christopherson, K.S., et al., Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell, 2005. 120(3): p. 421-33.
- Eroglu, C., et al., Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell, 2009. 139(2): p. 380-92.
- Risher, W.C., et al., Thrombospondin receptor alpha2delta-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol, 2018. 217(10): p. 3747-3765.
- Kucukdereli, H., et al., Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A, 2011. 108(32): p. E440-9.
- Risher, W.C., et al., Astrocytes refine cortical connectivity at dendritic spines. Elife, 2014. 3.
- Singh, S.K., et al., Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1alpha and NL1 via Hevin. Cell, 2016. 164(1-2): p. 183-196.
- Jones, E.V., et al., Astrocytes control glutamate receptor levels at developing synapses through SPARC-beta-integrin interactions. J Neurosci, 2011. 31(11): p. 4154-65.
- Mauch, D.H., et al., CNS synaptogenesis promoted by glia-derived cholesterol. Science, 2001. 294(5545): p. 1354-7.
- Goritz, C., D.H. Mauch, and F.W. Pfrieger, Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci, 2005. 29(2): p. 190-201.
- Gomez-Casati, M.E., et al., Nonneuronal cells regulate synapse formation in the vestibular sensory epithelium via erbB-dependent BDNF expression. Proc Natl Acad Sci U S A, 2010. 107(39): p. 17005-10.
- Hu, R., et al., Astrocyte-derived estrogen enhances synapse formation and synaptic transmission between cultured neonatal rat cortical neurons. Neuroscience, 2007. 144(4): p. 1229-40.
- Garrett, A.M. and J.A. Weiner, Control of CNS synapse development by {gamma}-protocadherin-mediated astrocyte-neuron contact. J Neurosci, 2009. 29(38): p. 11723-31.
- Feng, Z. and C.P. Ko, Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-beta1. J Neurosci, 2008. 28(39): p. 9599-609.
- Diniz, L.P., et al., Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia, 2014. 62(12): p. 1917-31.
- Diniz, L.P., et al., Astrocyte-induced synaptogenesis is mediated by transforming growth factor beta signaling through modulation of D-serine levels in cerebral cortex neurons. J Biol Chem, 2012. 287(49): p. 41432-45.
- Fossati, G., et al., Pentraxin 3 regulates synaptic function by inducing AMPA receptor clustering via ECM remodeling and beta1-integrin. EMBO J, 2019. 38(1).
- Allen, N.J., et al., Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature, 2012. 486(7403): p. 410-4.
- Farhy-Tselnicker, I., et al., Astrocyte-Secreted Glypican 4 Regulates Release of Neuronal Pentraxin 1 from Axons to Induce Functional Synapse Formation. Neuron, 2017. 96(2): p. 428-445 e13.
- Blanco-Suarez, E., et al., Astrocyte-Secreted Chordin-like 1 Drives Synapse Maturation and Limits Plasticity by Increasing Synaptic GluA2 AMPA Receptors. Neuron, 2018. 100(5): p. 1116-1132 e13.
- Chih, B., H. Engelman, and P. Scheiffele, Control of excitatory and inhibitory synapse formation by neuroligins. Science, 2005. 307(5713): p. 1324-8.
- Stogsdill, J.A., et al., Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature, 2017. 551(7679): p. 192-197.
- Kerr, K.S., et al., Glial wingless/Wnt regulates glutamate receptor clustering and synaptic physiology at the Drosophila neuromuscular junction. J Neurosci, 2014. 34(8): p. 2910-20.
- Nishida, H. and S. Okabe, Direct astrocytic contacts regulate local maturation of dendritic spines. J Neurosci, 2007. 27(2): p. 331-40.
- Carmona, M.A., et al., Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc Natl Acad Sci U S A, 2009. 106(30): p. 12524-9.
- Molofsky, A.V., et al., Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature, 2014. 509(7499): p. 189-94.
- Talmage, D.A., Mechanisms of neuregulin action. Novartis Found Symp, 2008. 289: p. 74-84; discussion 84-93.
- Walker, C.D., W.C. Risher, and M.L. Risher, Regulation of Synaptic Development by Astrocyte Signaling Factors and Their Emerging Roles in Substance Abuse. Cells, 2020. 9(2).
- Fuentes-Medel, Y., et al., Integration of a retrograde signal during synapse formation by glia-secreted TGF-beta ligand. Curr Biol, 2012. 22(19): p. 1831-8.
- Tjalsma, H., et al., Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol Mol Biol Rev, 2000. 64(3): p. 515-47.
- Tjalsma, H., et al., Proteomics of protein secretion by Bacillus subtilis: separating the "secrets" of the secretome. Microbiol Mol Biol Rev, 2004. 68(2): p. 207-33.
- Chung, W.S., N.J. Allen, and C. Eroglu, Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb Perspect Biol, 2015. 7(9): p. a020370.
- Allen, N.J. and C. Eroglu, Cell Biology of Astrocyte-Synapse Interactions. Neuron, 2017. 96(3): p. 697-708.
- Bosworth, A.P. and N.J. Allen, The diverse actions of astrocytes during synaptic development. Curr Opin Neurobiol, 2017. 47: p. 38-43.
- Baldwin, K.T. and C. Eroglu, Molecular mechanisms of astrocyte-induced synaptogenesis. Curr Opin Neurobiol, 2017. 45: p. 113-120.
- Xu, J., N. Xiao, and J. Xia, Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat Neurosci, 2010. 13(1): p. 22-4.
- Cahoy, J.D., et al., A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci, 2008. 28(1): p. 264-78.
- Chen, H., M.E. Herndon, and J. Lawler, The cell biology of thrombospondin-1. Matrix Biol, 2000. 19(7): p. 597-614.
- Mendus, D., et al., Thrombospondins 1 and 2 are important for afferent synapse formation and function in the inner ear. Eur J Neurosci, 2014. 39(8): p. 1256-67.
- Park, J., et al., Central Mechanisms Mediating Thrombospondin-4-induced Pain States. J Biol Chem, 2016. 291(25): p. 13335-48.
- Zhang, S., et al., Constitutive expression of pentraxin 3 (PTX3) protein by human amniotic membrane cells leads to formation of the heavy chain (HC)-hyaluronan (HA)-PTX3 complex. J Biol Chem, 2014. 289(19): p. 13531-42.
- Lu, L., et al., Common and rare variants of the THBS1 gene associated with the risk for autism. Psychiatr Genet, 2014. 24(6): p. 235-40.
- Barbati, E., et al., Influence of pentraxin 3 (PTX3) genetic variants on myocardial infarction risk and PTX3 plasma levels. PLoS One, 2012. 7(12): p. e53030.
- Cunha, C., O. Kurzai, and A. Carvalho, PTX3 deficiency and aspergillosis. N Engl J Med, 2014. 370(17): p. 1666-7.
- Bonacina, F., et al., Pentraxin 3 deficiency protects from the metabolic inflammation associated to diet-induced obesity. Cardiovasc Res, 2019. 115(13): p. 1861-1872.
- Lopez-Murcia, F.J., B. Terni, and A. Llobet, SPARC triggers a cell-autonomous program of synapse elimination. Proc Natl Acad Sci U S A, 2015. 112(43): p. 13366-71.
- Clarke, L.E. and B.A. Barres, Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci, 2013. 14(5): p. 311-21.
- Seddighi, S., et al., SPARCL1 Accelerates Symptom Onset in Alzheimer's Disease and Influences Brain Structure and Function During Aging. J Alzheimers Dis, 2018. 61(1): p. 401-414.
- Dowling, C. and N.J. Allen, Mice Lacking Glypican 4 Display Juvenile Hyperactivity and Adult Social Interaction Deficits. Brain Plast, 2018. 4(2): p. 197-209.
- O'Brien, R.J., et al., Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron, 1999. 23(2): p. 309-23.
- O'Brien, R., et al., Synaptically targeted narp plays an essential role in the aggregation of AMPA receptors at excitatory synapses in cultured spinal neurons. J Neurosci, 2002. 22(11): p. 4487-98.
- Xu, D., et al., Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron, 2003. 39(3): p. 513-28.
- Sia, G.M., et al., Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron, 2007. 55(1): p. 87-102.
- Chang, M.C., et al., Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nat Neurosci, 2010. 13(9): p. 1090-7.
- Gu, Y., et al., Obligatory role for the immediate early gene NARP in critical period plasticity. Neuron, 2013. 79(2): p. 335-46.
- Pelkey, K.A., et al., Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron, 2015. 85(6): p. 1257-72.
- Lee, S.J., et al., Presynaptic Neuronal Pentraxin Receptor Organizes Excitatory and Inhibitory Synapses. J Neurosci, 2017. 37(5): p. 1062-1080.
- Tenorio, J., et al., Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis, 2014. 9: p. 138.
- Buosi, A.S., et al., Heterogeneity in Synaptogenic Profile of Astrocytes from Different Brain Regions. Mol Neurobiol, 2018. 55(1): p. 751-762.
- Chai, H., et al., Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron, 2017. 95(3): p. 531-549 e9.
- Lanjakornsiripan, D., et al., Layer-specific morphological and molecular differences in neocortical astrocytes and their dependence on neuronal layers. Nat Commun, 2018. 9(1): p. 1623.
- Morel, L., et al., Molecular and Functional Properties of Regional Astrocytes in the Adult Brain. J Neurosci, 2017. 37(36): p. 8706-8717.
- Boisvert, M.M., et al., The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep, 2018. 22(1): p. 269-285.
- Holt, L.M., S.T. Stoyanof, and M.L. Olsen, Magnetic Cell Sorting for In Vivo and In Vitro Astrocyte, Neuron, and Microglia Analysis. Curr Protoc Neurosci, 2019. 88(1): p. e71.
- Yu, X., J. Nagai, and B.S. Khakh, Improved tools to study astrocytes. Nat Rev Neurosci, 2020. 21(3): p. 121-138.
- Batiuk, M.Y., et al., Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun, 2020. 11(1): p. 1220.
- Ben Haim, L. and D.H. Rowitch, Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci, 2017. 18(1): p. 31-41.
- John Lin, C.C., et al., Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci, 2017. 20(3): p. 396-405.
- Lee, E. and W.S. Chung, Glial Control of Synapse Number in Healthy and Diseased Brain. Front Cell Neurosci, 2019. 13: p. 42.
- Jung, Y.J. and W.S. Chung, Phagocytic Roles of Glial Cells in Healthy and Diseased Brains. Biomol Ther (Seoul), 2018. 26(4): p. 350-357.
- Stevens, B., et al., The classical complement cascade mediates CNS synapse elimination. Cell, 2007. 131(6): p. 1164-78.
- Allen, N.J., Astrocyte regulation of synaptic behavior. Annu Rev Cell Dev Biol, 2014. 30: p. 439-63.
- Nguyen, J.V., et al., Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci U S A, 2011. 108(3): p. 1176-81.
- Villa, A., S. Della Torre, and A. Maggi, Sexual differentiation of microglia. Front Neuroendocrinol, 2019. 52: p. 156-164.
- VanRyzin, J.W., et al., Microglia and sexual differentiation of the developing brain: A focus on extrinsic factors. Glia, 2020. 68(6): p. 1100-1113.
- Crespo-Castrillo, A., L.M. Garcia-Segura, and M.A. Arevalo, The synthetic steroid tibolone exerts sex-specific regulation of astrocyte phagocytosis under basal conditions and after an inflammatory challenge. J Neuroinflammation, 2020. 17(1): p. 37.
- Chung, W.S., et al., Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature, 2013. 504(7480): p. 394-400.
- Scott-Hewitt, N., et al., Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J, 2020: p. e105380.
- Bialas, A.R. and B. Stevens, TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci, 2013. 16(12): p. 1773-82.
- Schafer, D.P., et al., Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 2012. 74(4): p. 691-705.
- Sellgren, C.M., et al., Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci, 2019. 22(3): p. 374-385.
- Sarn, N., et al., Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Mol Psychiatry, 2020.
- Kim, H.J., et al., Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry, 2017. 22(11): p. 1576-1584.
- Penzes, P., et al., Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci, 2011. 14(3): p. 285-93.
- Lepeta, K., et al., Synaptopathies: synaptic dysfunction in neurological disorders - A review from students to students. J Neurochem, 2016. 138(6): p. 785-805.
- Kaminsky, N., et al., Connecting Malfunctioning Glial Cells and Brain Degenerative Disorders. Genomics Proteomics Bioinformatics, 2016. 14(3): p. 155-165.
- Oksanen, M., et al., Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell Mol Life Sci, 2019. 76(14): p. 2739-2760.
- Henstridge, C.M., M. Tzioras, and R.C. Paolicelli, Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration. Front Cell Neurosci, 2019. 13: p. 63.
- Vainchtein, I.D., et al., Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science, 2018. 359(6381): p. 1269-1273.
- Delmas, C. and E. Dalmas, IL-33 Deals with the Gray Matter. Immunity, 2018. 48(3): p. 484-486.
- Vainchtein, I.D. and A.V. Molofsky, Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci, 2020. 43(3): p. 144-154.
- Pomeshchik, Y., et al., Interleukin-33 treatment reduces secondary injury and improves functional recovery after contusion spinal cord injury. Brain Behav Immun, 2015. 44: p. 68-81.
- Ackerman, S.D., et al., 2020.
- Henley, J.M. and K.A. Wilkinson, Synaptic AMPA receptor composition in development, plasticity and disease. Nat Rev Neurosci, 2016. 17(6): p. 337-50.
- Jia, Z., et al., Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron, 1996. 17(5): p. 945-56.
- Traynelis, S.F., et al., Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev, 2010. 62(3): p. 405-96.
- Baldwin, K.T. and C. Eroglu, Astrocytes "Chordinate" Synapse Maturation and Plasticity. Neuron, 2018. 100(5): p. 1010-1012.
- Webb, T.R., et al., X-linked megalocornea caused by mutations in CHRDL1 identifies an essential role for ventroptin in anterior segment development. Am J Hum Genet, 2012. 90(2): p. 247-59.
- De Pitta, M., N. Brunel, and A. Volterra, Astrocytes: Orchestrating synaptic plasticity? Neuroscience, 2016. 323: p. 43-61.
- Haber, M., L. Zhou, and K.K. Murai, Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J Neurosci, 2006. 26(35): p. 8881-91.
- Schiweck, J., B.J. Eickholt, and K. Murk, Important Shapeshifter: Mechanisms Allowing Astrocytes to Respond to the Changing Nervous System During Development, Injury and Disease. Front Cell Neurosci, 2018. 12: p. 261.
- Koeppen, J., et al., Functional Consequences of Synapse Remodeling Following Astrocyte-Specific Regulation of Ephrin-B1 in the Adult Hippocampus. J Neurosci, 2018. 38(25): p. 5710-5726.
- Nguyen, A.Q., et al., Astrocytic Ephrin-B1 Controls Synapse Formation in the Hippocampus During Learning and Memory. Front Synaptic Neurosci, 2020. 12: p. 10.
- Nikolakopoulou, A.M., et al., Astrocytic Ephrin-B1 Regulates Synapse Remodeling Following Traumatic Brain Injury. ASN Neuro, 2016. 8(1): p. 1-18.
- van Deijk, A.F., et al., Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia, 2017. 65(4): p. 670-682.
- Valenza, M., et al., Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington's disease. Cell Death Differ, 2015. 22(4): p. 690-702.
- Kacher, R., et al., CYP46A1 gene therapy deciphers the role of brain cholesterol metabolism in Huntington's disease. Brain, 2019. 142(8): p. 2432-2450.
- Vance, J.E., Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech, 2012. 5(6): p. 746-55.
- Wang, H., et al., 2020.
- Jeong, W., et al., ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer's Disease. Mol Cells, 2019. 42(11): p. 739-746.
- Turrigiano, G., Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci, 2011. 34: p. 89-103.
- Thalhammer, A., et al., Alternative Splicing of P/Q-Type Ca(2+) Channels Shapes Presynaptic Plasticity. Cell Rep, 2017. 20(2): p. 333-343.
- Beattie, E.C., et al., Control of synaptic strength by glial TNFalpha. Science, 2002. 295(5563): p. 2282-5.
- Stellwagen, D., et al., Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci, 2005. 25(12): p. 3219-28.
- Ogoshi, F., et al., Tumor necrosis-factor-alpha (TNF-alpha) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp Neurol, 2005. 193(2): p. 384-93.
- Haydon, P.G. and M. Nedergaard, How do astrocytes participate in neural plasticity? Cold Spring Harb Perspect Biol, 2014. 7(3): p. a020438.
- Grassi, F., et al., TNF-alpha increases the frequency of spontaneous miniature synaptic currents in cultured rat hippocampal neurons. Brain Res, 1994. 659(1-2): p. 226-30.
- Pribiag, H. and D. Stellwagen, TNF-alpha downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J Neurosci, 2013. 33(40): p. 15879-93.
- Stellwagen, D. and R.C. Malenka, Synaptic scaling mediated by glial TNF-alpha. Nature, 2006. 440(7087): p. 1054-9.
- Araque, A., et al., Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci, 1999. 22(5): p. 208-15.
Figure 3. Glypican 4 and Neuronal Pentraxin 1. Astrocyte-derived Glypican 4 induces NP1 release from neurons through the PTPRδ receptor. NP1 stimulates AMPA receptors clustering on the postsynaptic terminal making the synapse functional.