In the recent years, the progress of international trade and travel has led to an increased risk of emerging infections. Around 75 percent of the pathogens causing these infections are of animal origin. Point-of-care tests (POCT) and point-of-need tests (PONT) have been established in order to directly provide accurate and rapid diagnostics at field level, the patient bed-side or at the site of outbreaks. These assays can help physicians and decision makers to take the right action without delay. Typically, POCT and PONT rely on genomic identification of pathogens or track their immunological fingerprint. Recently, protocols for metagenomic diagnostics in the field have been developed.
- point-of-care
- point-of-need
- diagnostics
- lateral flow assay
- isothermal amplification
- mobile laboratory
- field applicable diagnostics
1. Background
- Background
Many different microorganisms such as bacteria, viruses, fungi, or parasites exist in the environment. Most of them are important to maintain the ecosystem on earth, however, some of them are pathogenic and cause infectious diseases in both humans and animals. Infectious diseases are one of the major reasons for death especially in the low- and middle-income regions of the world [1].
While globalization and international travel progresses, the risk of emerging diseases is increasing. Due to global flight connections, people, goods, or traded animals can reach almost every location in the world within one or two days. Unfortunately, this time frame is shorter than the incubation periods of several important zoonotic and animal diseases with an emerging potential defined by the World Organization for Animal Health (OIE, Table 1,[2]).
Table 1. Incubation Times of Selected Important Zoonotic and Animal Diseases.
|
Pathogen |
Incubation Time (days) |
|
African swine fever virus |
5−21 |
|
Suid herpesvirus 1(Aujeszky’s disease) |
2−10 |
|
Classical swine fever virus |
2−14 |
|
Foot and mouth disease virus |
2−14 |
|
Influenza viruses |
1−4 |
|
Lumpy skin disease virus |
4−28 |
|
Ebola virus |
2−21 |
|
Marburg virus |
2−21 |
|
Middle East respiratory syndrome virus |
2−14 |
|
Rift valley fever virus |
2−6 |
|
Severe acute respiratory syndrome virus |
2−7 |
|
Hand, foot, and mouth disease viruses (Enterovirus) |
3−6 |
Around 75 percent of all new emerging diseases are zoonotic [3]. In many African countries, one of the causes leading to the emergence of pathogens is bush meat, since people living in rural areas depend on it as nutrition source. Not only the consumption but also the processing of the meat can lead to an infection [4]. Also, settlements in forests and deforestation are driving factors in the spread of infectious diseases as the habitats of pathogens´ vectors and reservoirs shrink and start to overlap with areas where domesticated animals and people live [5]. The danger of being infected by a zoonotic disease is not limited to low resource settings. In general, every person who lives or works in close contact to animals is at risk. This is particularly true for people living on a farm or together with pets but also for people in contact with wild or zoo animals [6]. Unfortunately, there is a lack of diagnostic capacity in many regions of the world. Thus, disease outbreaks may stay undetected for longer time and can spread through the population of the affected area. For instance, in 2013, only twelve countries in sub-Saharan Africa have laboratories accredited to international standards. Ninety-one percent of the 380 laboratories were located in South Africa [7]. These numbers show the urgent need for diagnostic capacity in sub-Saharan Africa.
Another example for the need of sophisticated diagnostics are natural disasters as they are often followed by infectious disease outbreaks. Normally an outbreak starts during the post-impact phase within the first four weeks after the disaster. Depending on the type of the catastrophe, the resulting disease differs. For instance, floods more often lead to mosquito-borne infections, while earthquakes are correlate with diseases occurring due to contaminated food or water sources [8]. The World Health Organization (WHO) defines a relatively-small number of diseases related to natural disasters. In contrast, interviews with experts involved in previous disaster situations revealed that there are several important pathogens, which are listed in Table 2 [9][10][9,10].
Table 2. Important Pathogens Related With Epidemic Outbreaks After Natural Disasters as Defined by Brock et al. [9] in Comparison to Pathogens Listed by the WHO [10].
|
. |
Brock et al. |
World Health Organization |
|
Bacteria |
Methicillin-resistant Staphylococcus aureus E.coli Pseudomonas aeruginosa Methicillin-sensitive Staphylococcus aureus Enterobacter Klebsiella Enterococcus faecalis Coagulase-negative Staphylococcus Streptococcus pyogenes Enterococcus faecium Serratia marcescens Streptococcus agalactiae Streptococcus viridans Acinetobacter baumanii Stenotrophomonas maltophilia |
Vibrio cholerae E.coli Clostridium tetani
|
|
Viruses |
Human immunodeficiency virus Hepatitis B virus Hepatitis C virus West Nile virus Human T-lymphotropic virus Cytomegalovirus West-Nile virus Dengue fever virus Epstein-Bar virus Parvovirus B19 Chikungunya virus |
Hepatitis A Hepatitis E Measles virus Dengue fever virus |
|
Other pathogens Plasmodia species Leptospira species Acute respiratory infections |
A profound surveillance system is one of the main factors in preventing the spread of infectious diseases [11], which can only be achieved with highly-advanced diagnostics. The current diagnostic approaches rely on centralized reference laboratories with high throughput, due to the need of complex and expensive devices. Point-of-care testing (POCT) describes the identification of pathogens near the patient with a fast turn-around time and the potential to immediately change in the health management [12]. While the term POCT is used for human patients and samples, point-of-need testing (PONT) has a broader meaning and also includes on-site testing of environment, animals, and food samples, although this term is not yet clearly defined. The WHO formulated the characteristics of POCT and PONT as affordable, sensitive, specific, user friendly, robust and rapid, equipment-free, and deliverable to those who need them (ASSURED criteria, [13]).
Examples for rapid diagnostics in the field are the POCTs for malaria and human immunodeficiency viruses (HIV). As a consequence of rapid testing, disease control as well as change in treatment and care was achieved particularly, in regions where stable electricity supply, highly trained personal, or specialized devices are not available [14]. Most POCT and PONT rely on immuno-techniques to detect antigens or antibodies, but methods for the identification of the pathogen at molecular level are on the rise [15]. In this review, we provide an overview of the latest field-applicable diagnostic methods and techniques as well as their implementation at point-of-care (POC) and point-of-need (PON).
2. Immunoassays for Identification of Pathogens and Antibodies
- Immunoassays for Identification of Pathogens and Antibodies
Immunoassays are based on binding of an antibody and an antigen to each other. The simplest and most used equipment-free assays are lateral flow immunoassays (LFIAs) as they are performed in a small disposable cartridge (Figure 1). LFIAs do not require pipetting or washing steps and can be performed by untrained personal. In addition, no cold chain is required. Labelling of the secondary antibodies is accomplished mostly by gold or silver nanoparticles. LFIAs are able to detect pathogen-specific antigens and/or antibodies within 10−30 minutes. Recently, multiplex LFIAs have been developed for detecting multiple targets in a single test [16].
Paper-based micro fluid devices (µPAD) are yet another development of the lateral flow technique. Here, the sample is guided by hydrophobic channels [17]. This design enables more sophisticated applications like the combination of multiple serological assays in a single device [18][19][18,19].
A critical drawback of this technology is the sensitivity and specificity, as lateral flow assays (LFA) highly depend on the labelling and binding affinity of the tested biomolecules [20]. A high concentration of molecules is necessary to achieve positive results. Therefore, LFA will produce a false negative result, when testing samples containing only few target molecules. Moreover, on some occasions sample preparation is necessary since the test design only allows liquid non-viscous samples. Also, quantification of targets is not possible using the LFA.
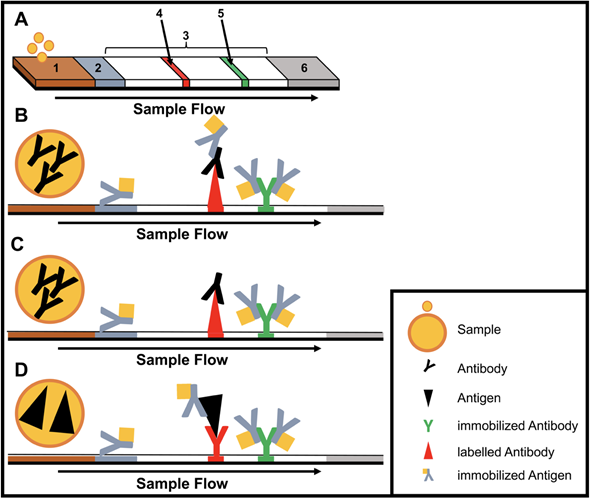
Figure 1. Structure and type of a lateral flow immunoassay. (A) Schematic design: 1, sample pad; 2, conjugate pad; 3, membrane; 4, test line; 5, control line; 6, adsorbent pad. (B) Indirect lateral flow immunoassay. (C) Competitive lateral flow immunoassay. (D) Sandwich lateral flow immunoassay. The principle of a lateral flow immunoassay (LFIA) is as follows—the sample is brought onto the sample pad and flows in the opposite direction by adsorption. While the sample passes the conjugate pad, a labelled target-specific antibody binds to the target molecule in the sample (antigen or antibody). Afterwards, the labelled antibody–target complex is immobilized to the membrane by a specific capture molecule (antibody or antigen) adhered to the membrane at the test line. The unbound labelled antibodies are captured at a control line by immobilized antibodies. In the case of a positive sample, the accumulation of the labelled antibodies leads to a coloration of both test lines. In the case of a negative sample, only the control line is colored. The adsorbent pad takes up the excess liquid. In the case of pathogens or targets, which are not suitable for the indirect or sandwich designed LFIAs, a competitive layout can be applied. This results in only one colored test line in case of a positive test.
3. Methods for the Identification of Pathogens at the Genomic Level
- Methods for the Identification of Pathogens at the Genomic Level
Nucleic acid amplification methods have the advantage of being highly sensitive as opposed to immunological assays due to the amplification step. DNA is amplified using cycling methods such as polymerase chain reaction (PCR) or isothermal amplification. In contrast to the PCR, isothermal amplification assays have the advantage of employing a constant reaction temperature for the amplification. This offers more utility in the field due to the use of portable heat sources and shorter run time in comparison to the cycle driven PCR [21]. Moreover, most isothermal methods are known to be resistant to inhibitors existing in complex samples like blood [22]. Several different methods for isothermal amplification have been developed in recent years (Table 3). From these methods, the two most evolving techniques are the loop-mediated isothermal amplification (LAMP, Figure 2) and the recombinase polymerase amplification (RPA, Figure 3). All features of both methods are compared in Table 4.
Table 3. Different Isothermal Amplification Techniques.
|
Method |
Reaction Temperature (°C) |
Time to Result (min) |
No. of Primers |
Probe |
|
Helicase-dependent amplification (HDA) |
37 |
60 |
2 |
− |
|
Rolling circle amplification (RCA) |
37 |
90 |
1,2 or > 2 |
+/- |
|
Recombinase polymerase Amplification (RPA) |
39−42 |
3−10 |
2 |
+ |
|
Nucleic acid sequence-based amplification (NASBA) |
41 |
90-120 |
2 |
+ |
|
Nicking enzyme amplification reaction (NEAR) |
60 |
2−5 |
2 |
+/- |
|
Loop-mediated isothermal amplification (LAMP) |
60−65 |
60 |
6 |
+/- |
+ use of probe for real-time detection possible; +/- use of probe possible, but usually not applied; - use of probe not possible.
Table 4. Features of Loop-Mediated Isothermal Amplification (LAMP) and Recombinase Polymerase Amplification (RPA).
|
Feature |
LAMP |
RPA |
|
Isothermal |
+ |
+ |
|
Visual read-out |
+ |
|
|
Portable heat source |
+ |
+ |
|
Easy to implement in field applications |
+ |
+ |
|
Fast result |
|
+ |
|
Pair of primers |
|
+ |
|
Simple assay design |
|
+ |
|
Highly resistant to inhibitors |
|
+ |
|
Long storage of reagents at room temperature |
|
+ |
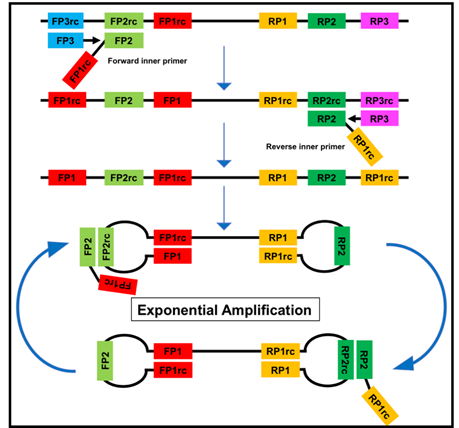
Figure 2. The loop-mediated isothermal amplification (LAMP) is based on a so called auto-cycling strand-displacement DNA synthesis using a strand-displacing DNA polymerase and two pairs of target-specific primers. The inner primers contain two sequences each corresponding to the sense and anti-sense (rc) strand of the target DNA, respectively, linked with a TTTT spacer. The inner primers initiate a complementary synthesis of the target DNA while the outer primers (FP1 and RP3) initiate a strand-displacement synthesis resulting in the release of single-stranded DNA linked by inner primers. The single-stranded DNA forms stem-loops by self-annealing of the corresponding sequences and acts as template for exponential amplification [23].
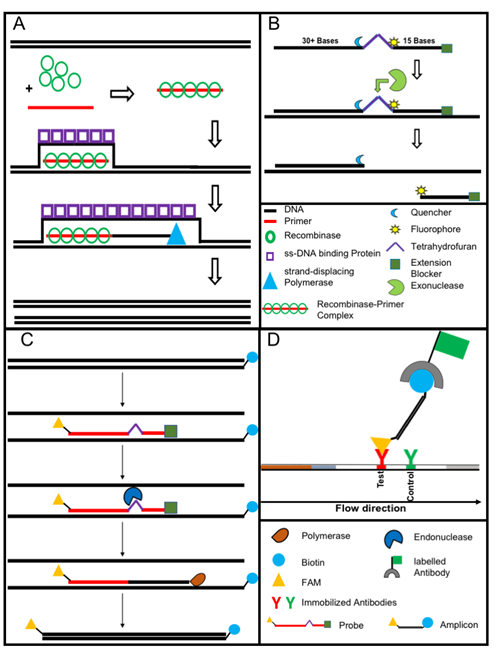
Figure 3. The recombinase polymerase amplification (A) employs a recombinase enzyme, a single-stranded binding protein, a strand-displacing (ss-DNA) polymerase, and a pair of target-specific primers. The primers form together with the recombinase enzyme the recombinase—primer complex which allows them to invade the double-stranded helix of the target DNA at the respective recognition site. The strand-displacing polymerase elongates the primers while the ss-DNA binding protein stabilizes the displaced strand in order to prevent self-annealing and ejection of the primers by branch migration. Real-time detection of amplification (B) is achieved by a sequence specific probe consisting out of a tetrahydrofuran abasic–site mimic (THF) flanked by fluorophore and quencher-labelled nucleotides as well as an extension blocker at the 3´ end. As the probe pairs with the complimentary sequence, a double-strand specific exonuclease slices the THF and the fluorophore is dissociated resulting in a signal [24]. Detection of RPA amplicon is in a lateral flow format (C and D). The use of a 5`end Biotin-labelled reverse primer in the RPA reaction leads to an antigenic-tagged reverse strand. When the 5-Carboxyfluorescein (FAM)-labelled probe binds to the complimentary strand a double-strand specific endonuclease slices the THF. The extension blocker is released and the remaining oligonucleotide can act as primer resulting in a FAM- and biotin-labelled amplicon (C), which can be detected in a lateral flow sandwich format (D,[25]).
3.1. Equipment-Free Nucleic Acid Amplification
The implementation of equipment-free nucleic acid amplification at POC and PON is extremely challenging. Although there are equipment-free LAMP assays, they still need a heating source [26][27][26,27]. Nevertheless, to overcome the required power supply for the LAMP reaction, LaBarre et al. developed a portable heating cartridge based on exothermal-reaction and engineered phase-change material. Heat is generated by the reaction of calcium oxide and water. To keep the temperature in the optimal range, a fat-based compound with a specific heat capacity and melting point (65 °C) is applied. Beside LAMP, this cartridge is also suitable for other isothermal reaction like RPA or the nicking enzyme amplification reaction NEAR [28].
Nucleic acid lateral flow (immuno-) assays (NALFAs/NALFIAs) provided a simple and inexpensive read-out of the different amplification methods [29][29]. These assays are mainly based on the capture of tagged amplicons or detection of amplicons in a sandwich format [30]. The use of the lateral flow technique removes the need for sophisticated devices (e.g., a fluorescence reader). In some studies, isothermal amplifications were shown to operate on paper-based devices. While paper-based RCA [31] works on room temperature without equipment. RPA was also conducted equipment-free using the body heat for incubation combined with a lateral flow-based detection of the tagged amplicons. The principle of RPA lateral flow detection is shown in Figure 3C,D, [32].
The bottleneck of the molecular diagnostics is the nucleic acid extraction as high quality DNA is needed for both DNA amplification assays and sequencing. Moreover, the sensitivity of diagnostic method is questioned if the right extraction method is not implemented. Despite many breakthroughs and full automation of the DNA and RNA extraction, very few studies have tried to set up a field-applicable extraction procedure. The most common extraction procedure relies on the binding of the nucleic acid to a silica gel membrane after a lysis step using a cocktail of chaotropic salts and proteinases [33]. In such case, a high-speed centrifuge is necessary. Magnetic bead-based purification methods omit the need for a centrifugation step, but several pipetting steps are still essential and total hands-on time exceeds 60 minutes [34]. Recently, a simple one-step purification protocol was applied to the extraction nucleic acid of paratuberculosis from fecal samples [35], Leishmania from blood and skin [36][37][36,37], Ebola from saliva [38], and rabies from tissue samples [39]. This method combined detergents and heat for the lysis step and magnetic beads to capture the inhibitors. The total run time was 15 minutes and one pipetting was applied as washing was not required. A quick extraction method, which incorporates detergent, proteinases, and heat, was used to obtain the RNA from hepatitis C [40] and Ebola viruses [41]. Another approach based on two steps of alkaline lysis then neutralization was used for DNA collection from skin samples of Buruli ulcer patients [42]. Despite the availability of rapid extraction techniques, so far, they have not been widely used. Another important issue is the demand for non-invasive samples for diagnostics to avoid the hassle of the need for a highly-qualified person to collect complex samples like blood and cerebrospinal fluid at PON [43].
4. Metagenomic Diagnostics as a Tool for Outbreak Identification
- Metagenomic Diagnostics as a Tool for Outbreak Identification
Metagenomic diagnostics is the identification of pathogens after sequencing of genomic material collected from suspected cases [44]. This approach is based on the sequencing and identification of either all the nucleic acids present in the sample (shotgun metagenomics) or a particular group of previously generated amplicons (targeted-amplicon sequencing) [45].
The molecular detection through PCR or isothermal amplification of a pathogen requires a known target nucleic acid sequence. Thus, in an outbreak of unknown cause, selection of the right molecular assay upon the clinical signs is challenging and a novel pathogen or variants of known infectious agent may remain undetected [46]. Metagenomic diagnostics overcomes this problem by sequencing and identifying all of the nucleic acids within the sample and thereby preparing for simpler diagnostic testing. Also, information about epidemiology and transmission route can be achieved [47]. However, the performance of this method strongly relies on the used genome database and depth of sequence analysis as well as on the amount of generated reads [48]. The major drawbacks of metagenomic diagnostics are the high costs per sequencing sample and the relatively long turnover times [47].
Sequencing methods require heavy and complex devices. However, the introduction of nanopore sequencing to the market by Nanopore Technologies allowed third-generation sequencing at PON due to small portable sequencing devices (Figure 4). The great utility of nanopore sequencing at PONf ne was proven by identification of pathogens direct from clinical samples [49] as well as during the surveillance for instance of Ebola and Zika outbreaks in West-Africa and South-America [50][51][50,51].
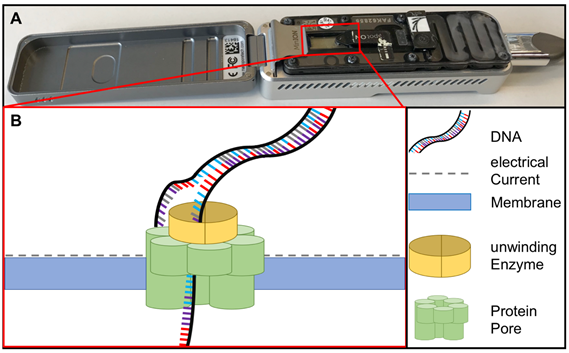
Figure 4. (A) MinION sequencing device (Oxford Nanopore technology, Cambridge, UK) and (B) the principle of nanopore sequencing. A protein pore is embedded in a membrane set under a certain electrical current. As the DNA molecule passes through the pore each nucleotide is recognized by the individual change caused in the voltage.