Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Maria Eleni Alexandrou and Version 2 by Catherine Yang.
Diabetic kidney disease (DKD) represents a major public health issue, currently posing an important burden on healthcare systems. Renin–angiotensin system (RAS) blockers are considered the cornerstone of treatment of albuminuric DKD. Aldosterone is a steroid hormone that is produced by the adrenal cortex and acts, together with other members of the steroid hormone family (cortisol and corticosterone), as a ligand of MRs.
- diabetic kidney disease (DKD)
- mineralocorticoid receptor antagonists (MRAs)
- spironolactone
- eplerenone
- finerenone
- apararenone
- esaxerenone
1. Physiological Role of Aldosterone and Mechanisms of Ligand-Specific Activation of Mineralocorticoid Receptors
MRs are nuclear receptors, structurally similar to glucocorticoid receptors (GRs), that are expressed in epithelial and non-epithelial tissues, serving as transcription factors of target genes that regulate cellular processes [1][25] (Figure 1). In epithelial cells of the distal nephron, where both MR and GR are expressed, aldosterone exerts its classical actions with regards to volume depletion and hyperkalemia by regulating sodium, chloride and potassium handling, through transcription of the epithelial sodium channel (ENaC), Cl−/HCO3− exchangers and ROMK channels [2][3][26,27]. Activation of MR in non-epithelial tissues, including cardiomyocytes, smooth muscle cells, fibroblasts and macrophages in heart tissue [4][5][28,29], and glomerular podocytes [6][30], monocytes [7][31] and mesangial cells [8][32] in the kidney tissue, targets expression of genes that are involved in tissue repair and results in excess inflammation and fibrosis [9][10][22,33]. Activation of GRs modulates transcription of responsive genes related to energy homeostasis, response to stress and control of inflammation [1][25]. While only cortisol binds to the GR, the MR has multiple ligands with high affinity, including both cortisol and aldosterone [1][9][22,25]. Despite the fact that cortisol reaches up to 1000-fold higher concentrations in several tissues, aldosterone is the primary physiological MR ligand in humans [11][34]. Pre-receptor conversion of cortisol by 11-beta-hydroxysteroid-dehydrogenase-2 (11βHSD2) to inactive cortisone is a key mechanism for maintaining MR selectivity in target tissues and regulating distinct MR functions in the heart and kidneys [10][12][33,35]. Differentiations in the concentrations of 11βHSD2 in each tissue represent the major modulator of this process, with the enzyme being abundantly expressed in distal tubular epithelial cells but not in cardiomyocytes, podocytes and macrophages, where cortisol is the primary physiological ligand of MRs [10][33].
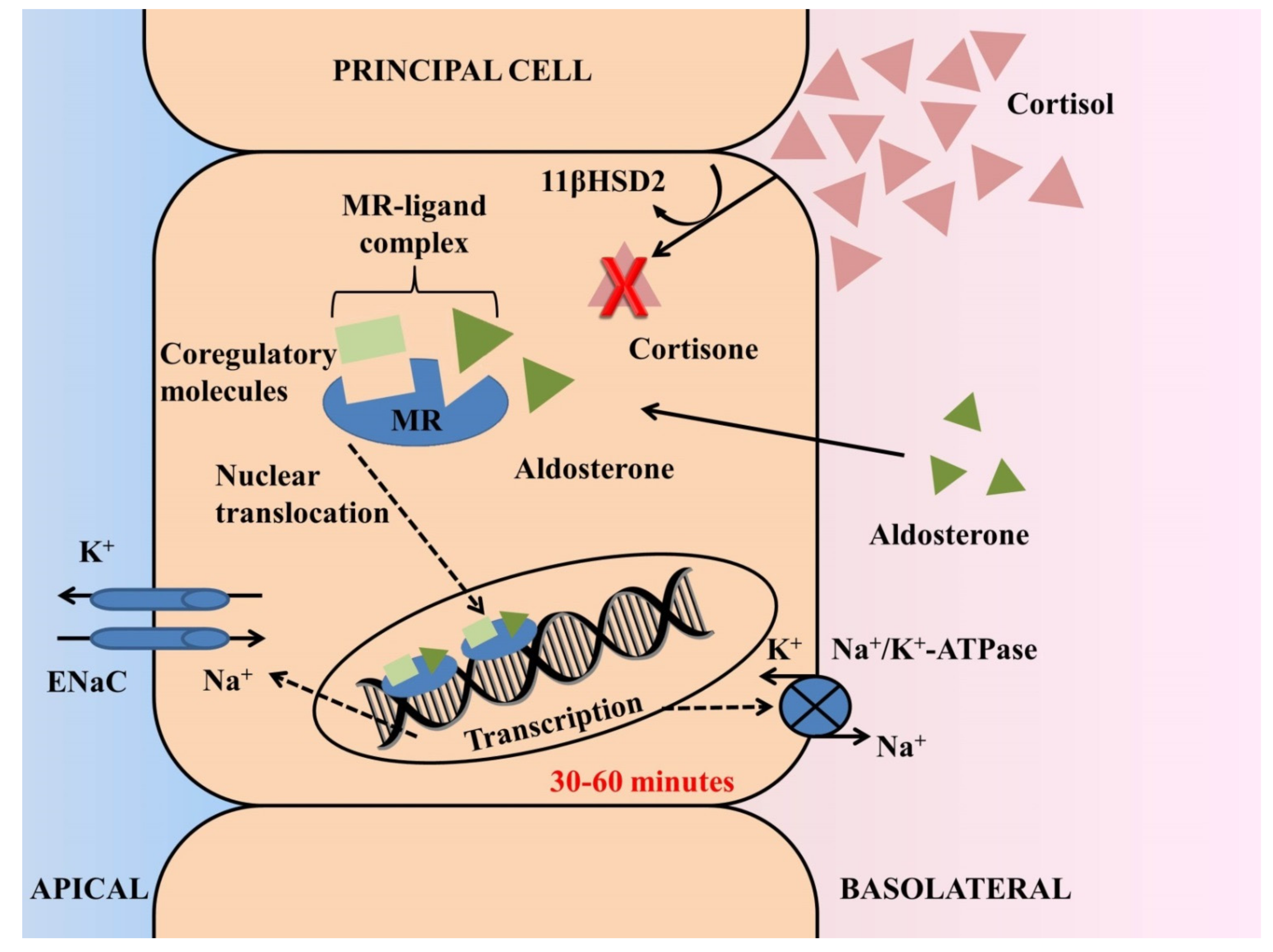
Figure 1. Actions of aldosterone through activation of the mineralocorticoid receptor (MR) in principal cells of the distal nephron. MR displays similar affinity for aldosterone and cortisol and ligand selectivity in the kidney is maintained by pre-receptor modulation. Despite the fact that cortisol reaches 100- to 1000-fold higher concentrations in several tissues, the 11-beta-hydroxysteroid-dehydrogenase-2 (11-HSD2) enzyme in tubular epithelial cells leads to conversion of cortisol to inactive cortisone, thereby rendering aldosterone the primary physiological MR ligand in the distal nephron. The genomic process begins after free diffusion of aldosterone through the cell membrane and fastening to the aldosterone ligand-binding domain of the MR. Upon ligand binding, transcription is dependent on recruitment of coregulatory proteins (coactivators or corepressors) and requires translocation of the MR-ligand complex into the nucleus. The final step of this process is upregulation of synthesis of the epithelial sodium channel (ENaC) at the apical membrane and of the Na/K–ATPase at the basolateral membrane, leading to renal sodium reabsorption and potassium secretion. In this classical genomic response, a latency of 30–60 min is expected after release or administration of aldosterone.
During the previous decade, the previously called nongenomic effects of aldosterone were elucidated. The genomic response is the process that includes all the classical steps of cell membrane diffusion of aldosterone: binding to the MR in the cytoplasm, translocation to the nucleus and activation of gene transcription [13][14][36,37]. This process results in an increase of ENaC concentration in epithelial cells (i.e., distal tubule, colon) and can be evidenced 30–60 min post aldosterone release/administration [15][38]. In addition to genomic actions, rapid effects of aldosterone have been described that cannot be explained by the traditional pathway, nor be blocked by inhibition of the transcriptional process molecules such as actinomycin D and aldosterone blockers, now considered to be mediated by MRs [16][39]. These rapid effects are associated with enhanced activity of the Na+-K+-2Cl− cotransporter and Na+-K+-ATPase in the heart, and of the Na+-H+ antiporter, the ENaC and Na+-K+-ATPase in the kidney, and are connected to subcellular trafficking [16][39].
In addition to the above, over the past years the crucial role of coregulatory molecules in mediating the genomic response to nuclear receptor activation has emerged [13][36]. Accumulated evidence suggests that upon ligand binding, the transcription of effector proteins is modulated via recruitment of coactivator or corepressor proteins according to distinct MR conformations induced by binding of different agonist ligands [1][13][25,36]. Ligand-selective peptides acting as potent antagonists of the MR-mediated transcription process were identified about a decade ago [13][36].
2. Aldosterone Breakthrough and Potential Mechanisms of Aldosterone-Induced Diabetic Kidney Disease and Cardiac Damage
In 10–53% of patients that initiate an ACEi/ARB, plasma aldosterone levels tend to rise again 6–12 months later, leaving them exposed to the deleterious proinflammatory and profibrotic effects of aldosterone in the kidneys, heart and vessels [2][17][18][26,40,41]. It has been speculated that this phenomenon of “aldosterone breakthrough” may represent a major cause of accelerated GFR decline in patients with type 1 diabetes and DKD and of poorer antiproteinuric response despite use of a single RAS blockade [10][33]. Inappropriate activation of MRs by aldosterone in podocytes, monocytes and mesangial cells in the kidney induces monocyte and macrophage infiltration [19][42] and promotion of glomerulosclerosis and interstitial fibrosis [20][43]. In the heart, overactivation of MRs promotes cardiac fibrosis, increased collagen synthesis and cardiac remodeling and hypertrophy [15][21][22][38,44,45]. An additional negative inotropic effect of aldosterone, counteracting the positive inotropic effect of angiotensin-II, has been also described [23][46].
Moreover, in the experimental model of streptozotocin-induced diabetes, a classical model of type 1 DM, overexpression of mRNA of MR, NADPH oxidase and collagen I/IV has been described, resulting in collagen deposition in glomerular and tubulointerstitial areas in the kidney [24][47]. Local production of aldosterone in mesangial cells induced by angiotensin-II, high glucose and LDL has been also associated with the pathogenesis of DKD through MR activation [25][48]. Administration of spironolactone was shown to block MR overexpression [24][47] and reduce collagen deposition in streptozotocin-induced diabetic rats [26][49]. Similarly, administration of spironolactone, eplerenone and finerenone in classical experimental models of DKD due to type 2 DM led to amelioration of glomerulosclerosis and macrophage infiltration [27][50], prevention of podocyte injury [28][51] and reduction of proteinuria and NGAL expression [29][52].
The crucial role of GR in progression of DKD has recently emerged. In an experimental model of streptozotocin-induced diabetes in podocyte GR knockout mice, the loss of podocyte GR resulted in enhanced Wnt signaling, higher expression of transforming growth factor-β and β-catenin and disturbed fatty acid metabolism, accompanied by histological evidence of worsened fibrosis, increased collagen deposition, as well as mesenchymal transition of the glomerular endothelium and glomerulomegaly [30][53]. Similarly, the loss of the endothelial GR has been shown to induce upregulation of Wnt/β-catenin signaling and to promote angiogenesis and mesenchymal transition in tubular epithelial cells in diabetic experimental models [31][32][54,55]. The above findings suggest the presence of a podocyte–endothelial cell crosstalk that is regulated by GR; this probably represents an additional mechanism in the development of diabetic nephropathy, on which more light should be shed in the future.