Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Fernando Remião and Version 2 by Dean Liu.
Cocaine is one of the most consumed stimulants throughout the world, as official sources report. It is a naturally occurring sympathomimetic tropane alkaloid derived from the leaves of Erythroxylon coca, which has been used by South American locals for millennia. Cocaine can usually be found in two forms, cocaine hydrochloride, a white powder, or ‘crack’ cocaine, the free base.
- sympathomimetics
- crack
- pharmacokinetics
- pharmacodynamics
- toxicity
- cocaine hydrochloride
- drug abuse
1. Introduction
Cocaine is a naturally occurring sympathomimetic alkaloid from the plant Erythroxylon coca that has been used as a stimulant, by chewing the leaves or brewing teas, in South America for over 5000 years. Cocaine was firstly isolated from the leaves in the mid-1800s and was at that time considered safe and used in toothache drops, nausea pills, energy tonics, and the original ‘Coca-Cola’ beverage [1][2][1,2]. Currently, it is found in one of two forms for (ab)use: Cocaine hydrochloride (also known as ‘coke’, ‘blow’ or ‘snow’), a fine white crystalline powder, which is soluble in water and consumed mainly through the intranasal route (‘sniffing’/‘snorting’), orally or intravenously; or as a free base (resulting from reaction of cocaine hydrochloride with ammonium or baking soda), commonly known as ‘crack cocaine’ or simply ’crack’, and typically consumed via inhalation (the solid mass is cracked into ‘rocks’ that are smoked, using glass or makeshift pipes) [2][3][2,3].
Cocaine abuse remains a significant public health problem with serious socio-economic consequences worldwide [4]. According to the most recent World Drug Report, 0.4% of the global population aged 15–64 reported cocaine use in 2019—this corresponds to approximately 20 million people [5]. The latest edition of the European Monitoring Centre for Drug and Drug Addiction (EMCDDA) Drug Report states that it remains the second most abused substance in the European Union, second only to cannabis [6]. Furthermore, despite the global COVID-19 pandemic, European authorities have intercepted at seaports growing amounts of cocaine in 2020 [5]. All the while, case reports detailing the harmful consequences of cocaine use abound [7][8][9][10][11][12][13][14][15][16][17][18][19][20][7,8,9,10,11,12,13,14,15,16,17,18,19,20].
2. Natural Occurrence and Chemical Characterisation of Erythroxylum coca
The coca shrub, from which cocaine is extracted, is a plant of the genus Erythroxylum that grows in Central and South America, and it has over 250 identified species, of which the two most important are E. coca and Erythroxylum novogranatense; however it is from E. coca Lam. var. coca (also referred to as ‘Bolivian’ or ‘Huanuco’ coca) that the majority of cocaine supply is extracted [21][22][21,22]. The coca plant presents large, thick, dark green leaves with an elliptical shape and a somewhat sharp apex, and has small red fruits [23]. Circa 18 different alkaloids can be found in the leaves of the coca plant, such as cinnamoylcocaine, tropacocaine, methylecgonine, benzoylecgonine (BE) and pseudotropine—all of these are significantly less euphoric and less toxic than cocaine (Figure 1) [21][22][21,22].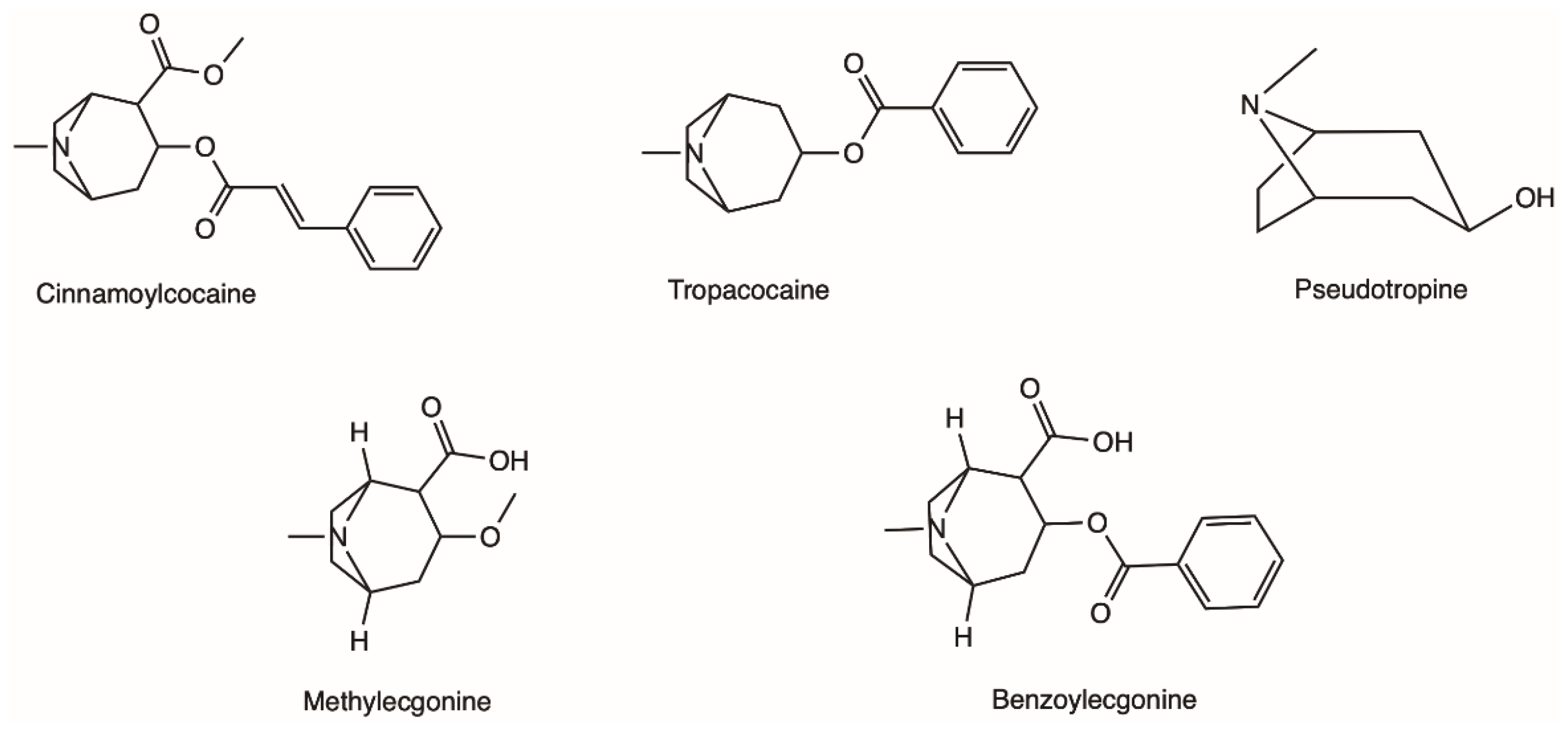
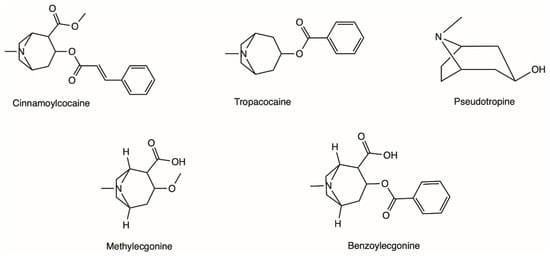
Figure 1.
Examples of different alkaloids that can be found in the leaves of the coca plant.
Coca leaves have been traditionally used by the indigenous Andean populations and were/are consumed mostly by chewing; coca leaves as a part of religious occasions and other celebrations by the Inca, as well as employed for medicinal purposes [22]. It was from the coca leaves that Albert Niemann first isolated cocaine in 1859–1860 [21][24][21,24]. A study from one hundred years later found that dry leaves of E. coca var. coca have around 6.3 mg of cocaine per gram of plant material [25].
One of the most employed methods of detection for cocaine are immunoassays, a fast method that allows a qualitative presumptive assessment of the drug in the biological matrix tested (e.g., blood, urine). However, as this type of test is subjected to a certain degree of false positive and false negative results, a confirmatory quantitative method should be employed afterwards [28][29][30][28,29,30]. Cocaine can be detected in both biological and non-biological matrices through chromatographic methods. A number of reports in the literature describe the detection of cocaine, and its metabolites, in human urine samples [31][32][33][34][35][31,32,33,34,35] but also in blood/plasma or hair [36][37][38][36,37,38]. A comparison of methodologies between gas chromatography coupled with mass spectrometry (GC-MS) and high-performance liquid chromatography (HPLC) for the analysis of human urine reported similar outcomes [39]. This allowed laboratories to resort to HPLC for sample examination without such a demanding pre-treatment. Kintz et al. first described a GC-MS method suitable for the simultaneous identification of the ‘crack’-specific metabolite, anhydroecgonine methyl ester (AEME), as well as the parent drug cocaine and additional metabolites (BE, ecgonine methyl ester (EME) and cocaethylene (CE)) in plasma, saliva, urine, sweat and hair samples [37]. This method was successfully applied to 1 mL samples of urine, saliva and plasma, 50 mg of hair, and sweat extracted from a sweat patch, yielding limits of detection for AEME of 1 ng/mL for the first three matrices (with a linearity range of 5–1500 ng/mL), 0.1 ng/mg for the hair (with a linearity range of 0.2–25 ng/mg) and 0.5 ng/patch (with a linearity range of 2–100 ng/patch). Recently, published research by Fernandez et al. described a validated method for the detection of cocaine and several of its metabolites in 0.5 mL of urine samples, resorting to a simple derivatization step following a solid phase extraction (SPE)—the method achieved low limits of quantification from 2.5 to 10 ng/mL by using GC-MS with electron ionization [40]. Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) was also successfully employed to detect cocaine and BE both in urine and oral fluid [41], as it was ultra-performance liquid chromatography coupled with tandem mass spectrometry (UPLC-MS/MS) in dried blood samples [42]. Of note, cocaine in blood/plasma samples may undergo spontaneous and enzymatic hydrolysis to its metabolite BE, if the samples are not treated with a pseudocholinesterase (PChE) inhibitor, such as sodium fluoride [24]. As such, samples suspected to contain cocaine should be adjusted to pH 5 with acetic acid and refrigerated at 4 °C or frozen to increase stability of the drug, although some degradation occurs along the time, even at −20 °C.
3. Physicochemical Properties of Cocaine and Analytical Methods for Identification
Cocaine is a tropane alkaloid with weak basic properties. In the free base form, cocaine is unionised and insoluble in aqueous medium, displaying a boiling point of 187 °C; while its ionised hydrochloride salt is readily dissolved in water and presents high stability at very high temperatures, as such, it does not volatilise in the smoke. Table 1 summarises a few of the physical and chemical properties of cocaine [26][27][26,27].Table 1. Physical and chemical properties of cocaine.
| Cocaine Form | Cocaine Hydrochloride | Cocaine Free Base |
|---|---|---|
| Other names it is known by | ‘coke’, ‘snow’, ‘blow’ | ‘crack’ |
| CAS | 53-21-4 | 50-36-2 |
| Molecular formula | C17H22ClNO4 | C17H21NO4 |
| Molecular weight | 339.8 g mol−1 | 303.35 g mol−1 |
| Boiling point | - | 187 °C |
| Melting point | 195 °C | 98 °C |
| Solubility in water | 2 g/mL | 1.7 × 10−3 g/mL |
| pKa; pKb (at 15 °C) | - | 8.61; 5.59 |
| Log P | - | 2.3 |
4. Pharmacodynamics
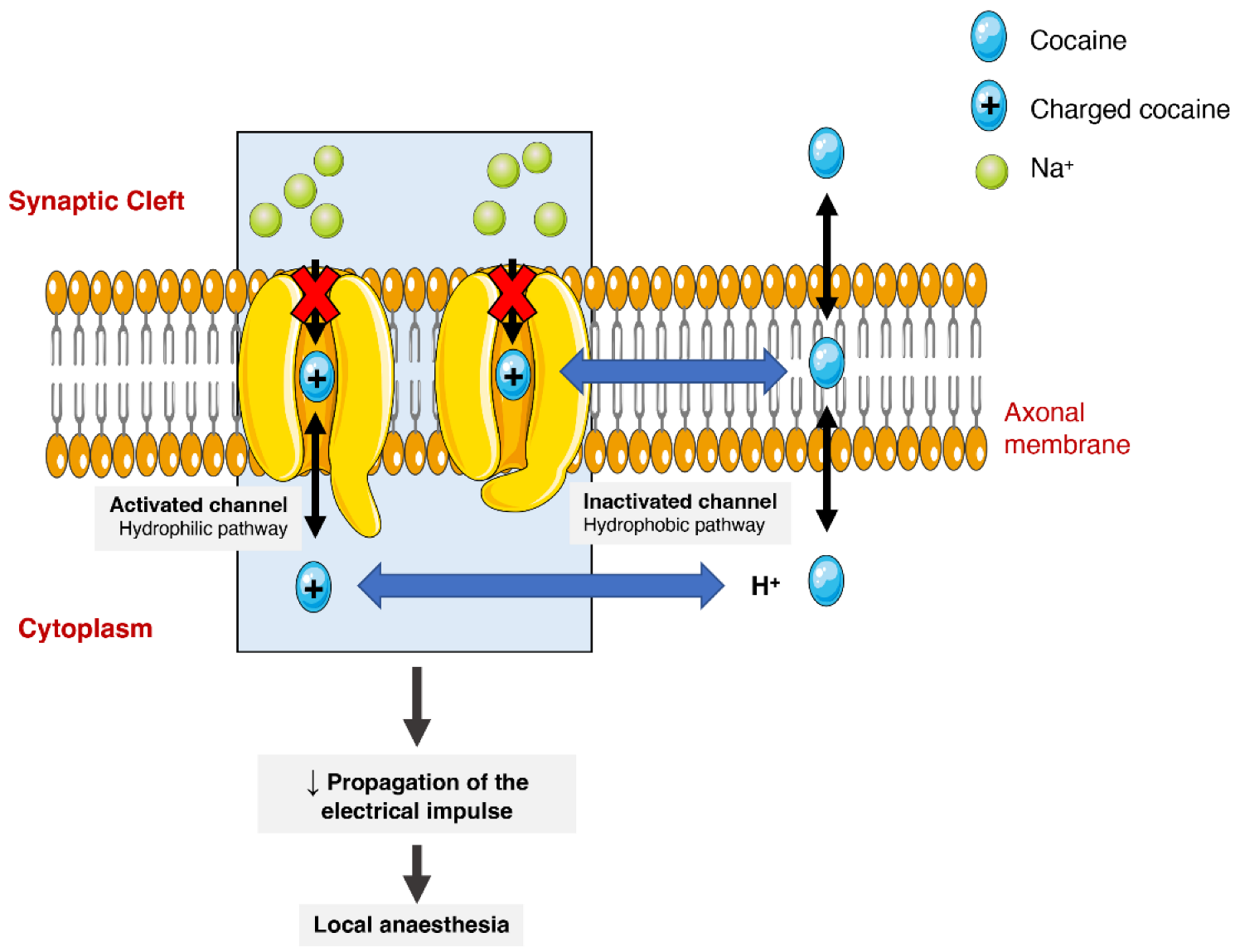
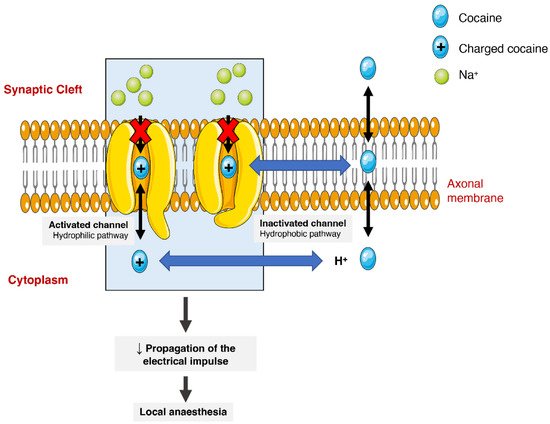
Cocaine has different pharmacodynamic properties that make possible its use as a local anaesthetic and as a sympathomimetic stimulant at the central nervous system (CNS). The anaesthetic action of cocaine is related to its capacity to block voltage-gated sodium channels by stabilizing these channels in an inactive state (Figure 23). The binding of cocaine to the channel’s pore prevents sodium from flowing through it into the cells and thus blocking the depolarization process and the propagation of the electrical impulses [1][24][43][1,24,75]. The current medical use is very limited as most countries consider it obsolete. It can still be used as a topical anaesthetic, which might be particularly useful for endoscopic sinus surgery, given its vasoconstrictive effects. There are, however, controversies related to the development of mild morbidities, such as hypertension and tachycardia [44][76].
Figure 23. Schematic representation of cocaine’s interaction with voltage-gated sodium channels. Cocaine enters the channels and binds to them by two pathways (hydrophilic and hydrophobic). In the hydrophobic pathway cocaine interacts with the sodium channel at the membrane level, alternatively in hydrophilic pathway, the cocaine is ionized in cytoplasm before the interaction. In both cases, the flow of sodium is blocked, which diminishes the propagation of electrical impulses and causes a local anaesthetic effect.
On the other hand, the psychoactive and sympathomimetic effects of cocaine derive from the blockade of presynaptic transporters responsible for the reuptake of serotonin, noradrenaline, and dopamine. In the case of the latter, the blockade of the presynaptic dopamine transporter (DAT) in the synaptic cleft causes an extracellular increase in dopamine with an overstimulation of the dopaminergic postsynaptic receptors, inducing the euphoric ‘rush’ [3][45][3,53]. Further mechanisms of tolerance at this level are responsible by the subsequent drop in the dopamine levels experienced as a dysphoric ‘crash’. A recent meta-analysis showed that chronic cocaine users display a significant reduction in dopamine receptors D2 and D3 in the striatum, the caudate and putamen brain regions, as well as a significantly increased availability of DAT all over the striatum [46][77].
When cocaine is consumed, an exacerbated dopaminergic activity along the mesocorticolimbic pathways occurs. Neurons from these pathways are located in the ventral tegmental area and project to other brain locations, including the nucleus accumbens [47][78]. This could explain why the drug has such an addictive potential, since it is well acknowledged that the nucleus accumbens may have an important role in the rewarding and addictive properties of cocaine and other drugs [48][79]. However, it should be mentioned that cocaine’s capacity to increase serotoninergic activity (which may induce seizures) could also contribute to the drug’s addictive potential [49][50][80,81]. Figure 34 schematically represents cocaine’s pharmacodynamic action over the monoaminergic system.
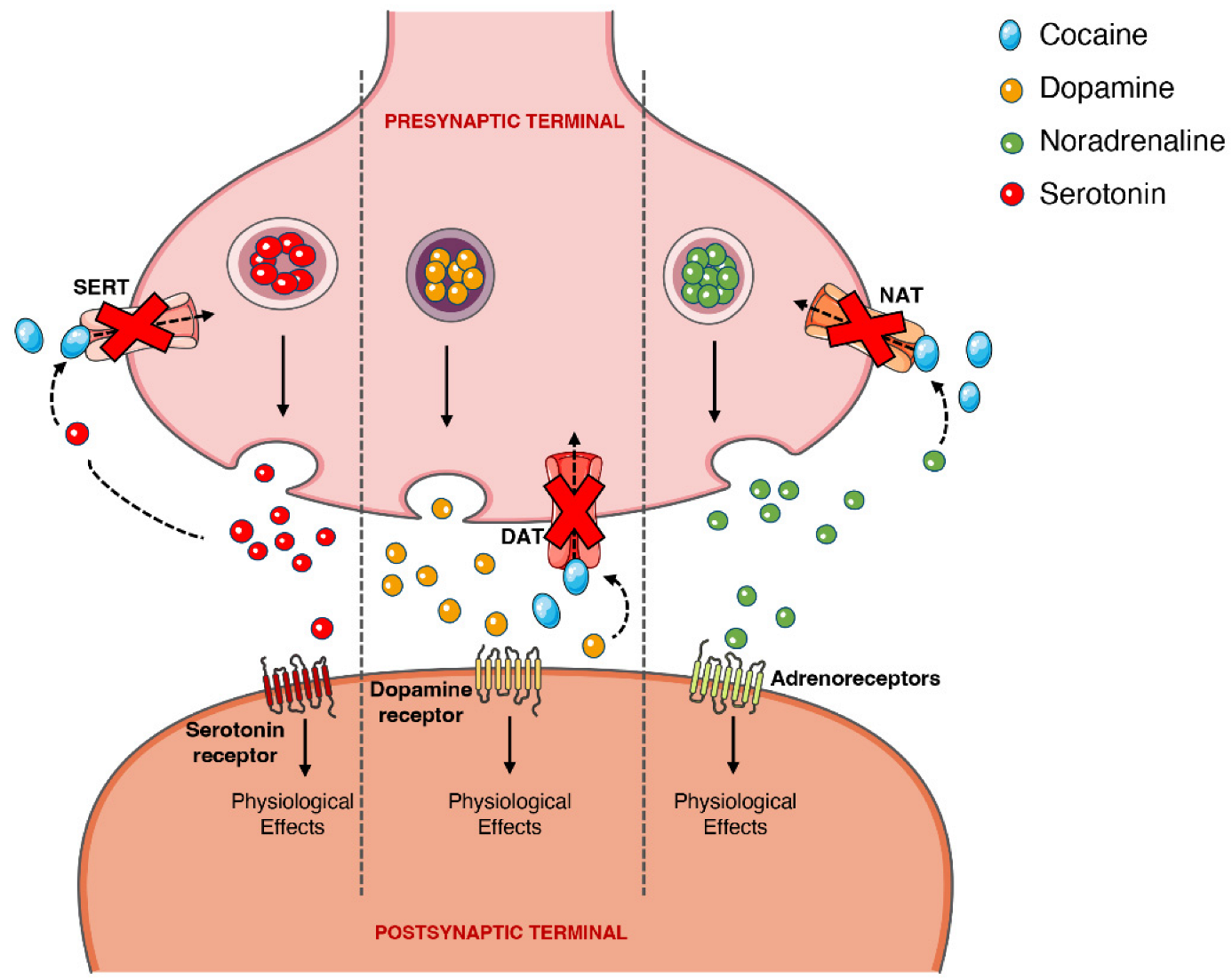
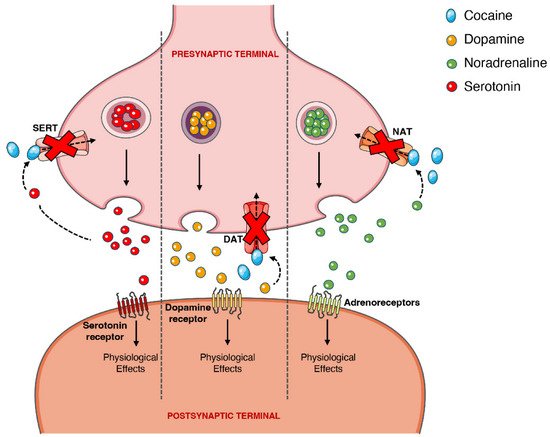
Figure 34. Schematic representation of cocaine’s pharmacodynamics at the noradrenergic, serotonergic or dopaminergic synapse. Cocaine acts by blocking the presynaptic transporters of dopamine, serotonin and noradrenaline, preventing the reuptake of the neurotransmitters into the presynaptic terminal, which will cause intense and prolonged stimulation of the postsynaptic receptors. DAT, dopamine transporter; NAT, noradrenaline transporter; SERT, serotonin transporter.
The sympathomimetic properties of cocaine are related to the above-mentioned inhibition of noradrenaline reuptake via noradrenaline transporter (NAT). Because cocaine impedes this reuptake of noradrenaline, and thus increases its availability, there will be an increase in the stimulation of the α- and β-adrenergic receptors, and an augmented adrenergic response—which relates to the marked vasoconstrictive properties of the drug (responsible for a few of the cardiotoxic effects) [2][51][52][2,82,83].
Additionally, cocaine also has the capacity to directly target adrenergic, N-methyl-D-aspartate (NMDA), and sigma and kappa opioid receptors. Cocaine affects NMDA receptors, as exposure to the drug modulates (for greater or lesser) receptor subunit expression, alters receptor distribution in the synapse, and influences the crosstalk of the NMDA receptor with the dopaminergic receptor D1, in different brain areas, for example, the nucleus accumbens, the ventral tegmental area and the prefrontal cortex [53][84]. Lastly, cocaine acts directly over the sigma opioid receptors, binding with greater affinity to the σ1 receptor than to the σ2 receptor; agonism at the σ1 by cocaine partially mediates the hyperlocomotion and seizures, and these receptors are paramount in the establishment of cocaine-induced conditioned place preference in mice [45][51][54][53,82,85].
Recently, it has been suggested that the pharmacological action of cocaine over DAT may not be as simple as the sole inhibition of the transporter’s reuptake function, as its behaviour is distinct from other DAT inhibitors of equal or greater potency (with matched capacity for crossing the BBB) and resembles methylphenidate (a norepinephrine-dopamine reuptake inhibitor that also induces the release of synaptic dopamine). As such, it was hypothesised that, similar to amphetamines, cocaine functions as a negative allosteric modulator of DAT (i.e., a DAT ‘inverse agonist’), altering transporter function and reversing transport direction [55][86]. However, more research is necessary in this area to further clarify cocaine pharmacodynamics.