Chronic hepatitis B virus (HBV) infection affects 292 million people worldwide and is associated with a broad range of clinical manifestations including cirrhosis, liver failure, and hepatocellular carcinoma (HCC). Despite the availability of an effective vaccine HBV still causes nearly 900,000 deaths every year. Current treatment options keep HBV under control, but they do not offer a cure as they cannot completely clear HBV from infected hepatocytes. The recent development of reliable cell culture systems allowed for a better understanding of the host and viral mechanisms affecting HBV replication and persistence. Recent advances into the understanding of HBV biology, new potential diagnostic markers of hepatitis B infection, as well as novel antivirals targeting different steps in the HBV replication cycle are summarized in this review article.
- HBV
- HBV RNA
- HBcrAg
- novel viral markers
- direct-acting antivirals
- Current Global Status
1. Current Global Status
Almost 60 years after the discovery of Australian antigen (AuAg) and more than 30 years after the approval of the first vaccine, hepatitis B virus (HBV) infection remains the most common chronic infectious disease in humans corresponding to 292 million people globally [1]. According to the World Health Organization (WHO), at least 2 billion people live with serological evidence of past or present infection with HBV, and nearly 900,000 deaths every year are caused by HBV-related complications, including hepatic failure and hepatocellular carcinoma (HCC) [2]. The global incidence of cirrhosis and HCC associated with hepatitis B is around 30% and 45%, respectively, with the proportion as high as 60% and 80% in highly endemic areas (East Asia, Africa) [3]. Importantly, up to 72 million people worldwide are coinfected with hepatitis delta virus (HDV), which is associated with more rapid progression to cirrhosis, a higher incidence of HCC, and increased mortality compared to chronic HBV monoinfection [4].
The epidemiology of HBV infection has always been determined by the detection of the hepatitis B surface antigen (HBsAg) in the general population. In concordance to these data the geographic prevalence of HBV infection is classified into three regions of high (>8%, East Asia, Africa), medium (2–8%, Mediterranean, Eastern Europe), and low (<2%, North America, Western Europe) endemicity[1]. Over the past few years, the epidemiology of HBV infection is changing due to the introduction of the universal vaccination program, hepatitis B screening programs as well as through the human migration between high and low-risk areas [5]. Currently, the complete vaccination schedule in healthy individuals consists of three doses of vaccine and according to the clinical trials the first dose should be delivered as soon as possible after birth (within 24 h) to prevent perinatal HBV infection and induce immunity against HBV. Despite the WHO recommendations, by 2015 only 55% of European countries reported offering Hepatitis B Birth dose (HepB-BD) as part of their National Vaccine Plan, while in remaining countries infants received the first dose of vaccine at 2 months of age or even later [6]. Therefore, the current main route of HBV is vertical, or mother to child transmission (MTCT) that is responsible for approximately 50% of the global disease burden [7]. Nevertheless, other sources such as sexual contact, inadequate sterilization of health care instruments, administration of contaminated blood products, as well as intravenous drug use also remain important modes of transmission. Moreover, special attention should be paid to highly endemic areas (Asia Pacific, sub-Saharan African) where the vertical transmission remains predominant route of infection. The majority of HBV carriers in these regions are infected at birth or during their first 5 years of life, when the risk of progression to chronicity is high [8–10][8][9][10]. This phenomenon is caused by the low coverage of the HBV birth dose, concerns about appropriate vaccine storage, unsuccessful prophylaxis against HBV, as well as high incidence of home births [8,10]. Another reason for HBV infection spreading is the poor or non-response to HBV vaccination. Despite the overall high efficiency of the vaccine, still, around 5% of individuals do not respond to the primary HBV series and the cause of this phenomenon remains unclear [11,12][11][12]. What should be also considered is the fact that the undetermined group of patients infected with HBV remains underdiagnosed by routine serological tests. These are individuals infected with escape mutants that lack HBsAg, or carriers with an occult infection which is defined as the persistence of viral genetic material in the liver in the absence of HBsAg in serum. According to the Global Hepatitis Report from 2017 only 9% of 257 million patients with a chronic infection have been diagnosed and knew of their status and in consequence as low as 1% of HBV carriers are properly treated worldwide. Furthermore, around 25% (15–40%) of those with chronic hepatitis B (CHB) if left untreated would develop cirrhosis, liver failure, or hepatocellular carcinoma (HCC). All these facts together with the lack of curative therapy against HBV were the cause of WHO implementation of the first global strategy to eliminate HBV infection as a public health threat by 2030 [13].
- HBV Virology
2. HBV Virology
The human HBV which belongs to the family Hepadnaviridae has one of the smallest viral genomes with a high organization in the enveloped viral particle. The circular, partially double-stranded DNA genome of HBV (relax circular DNA, rcDNA) is about 3.2 kb in size and comprises a complete coding minus strand (-) and a shorter noncoding plus strand (+) with a fixed 5′ end and a variable length 3’ end [14]. The minus strand of HBV encodes for highly overlapping four long open reading frames (ORFs: preC/C, P, preS/S, and X) that give rise to five RNA transcripts and seven viral proteins which include DNA polymerase (Pol), two nucleocapsid core proteins: hepatitis B core antigen (HBcAg) and hepatitis B e antigen (HBeAg), X protein (HBx), and three envelope proteins: large (L-), middle (M-), and small (S-) hepatitis B surface antigen (HBsAg) [15]. Unlike other DNA viruses, HBV has evolved an unusual genome replication strategy that allows for active replication without destroying the infected cells. Namely, the virus replicates through reverse transcription of an RNA intermediate, the pregenomic RNA (pgRNA), using the reverse-transcriptase activity of the viral polymerase which lacks the proofreading activity. Due to its replicative strategy and the complex nature of the genome, HBV exhibits an estimated 10-fold higher mutation rate than other DNA viruses [16]. Particularly, P gene overlaps with all other coding regions and any mutation in the polymerase gene may also affect the overlapping S gene influencing viral infectivity, liver disease severity, and also a response to antiviral treatment [16].
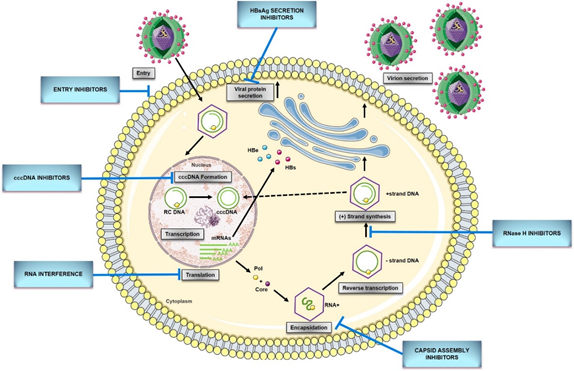
Figure 1. HBV replication cycle and new direct-acting antivirals for HBV. Entry: the replication cycle begins with the reversible binding of S-HBsAg to the glypican 5 (GPC5) protein [17], followed by specific binding between preS1 region of L-HBsAg and HBV entrance receptor—Sodium Taurocholate Cotransporting Peptide (NTCP) [18]. Upon the entrance, capsids are transported within the cytosol to the nucleus during which uncoating and release of the viral genome are initiated [14]. cccDNA formation: the molecular basis of rcDNA to cccDNA conversion remains unclear but it requires (1) the release of the viral polymerase covalently attached to the minus DNA strand by tyrosyl-DNA-phosphodiesterase 2 (TDP2) or its related proteins [19]; (2) the removal of the RNA primer from plus DNA strand by unrecognized enzymes; (3) cleavage of terminally redundant sequences (r) from the negative strand by structure-specific endonuclease 1 (FEN1) activity [20]; (4) the competition of positive DNA strand by the cellular replicative machinery: DNA polymerase κ [21] [21] or polymeraseα, δ, and ε [22], and DNA topoisomerase I and II [23]; (5) the ligation of both viral DNA strands by DNA ligase LIG I and LIG III [24]; (6) chromatinization involving histone chaperones, chromatin remodelers, transcription factors, and viral proteins [25]. Transcription and translation: cccDNA transcribes into all viral RNAs necessary for protein production and viral replication utilizing the cellular transcriptional machinery: 3.5-kb pregenomic RNA (pgRNA)/preC RNA and 2.4-kb, 2.1-kb, and 0.7-kb subgenomic RNA [15]. The pregenomic RNA encodes both the polymerase and core protein and works as a template for viral DNA replication. The three subgenomic RNAs encode envelope preS1 protein, preS2, and HBsAg proteins and the X protein [26]. The precore mRNA is translated into precore protein, which is processed at the N-terminal and C-terminal ends to HBeAg, a secretory protein. Pregenomic RNA is reverse transcribed into HBV DNA and also translated into core protein (HBcAg), which overlaps with HBeAg [27]. Encapsidation: the first step of HBV genome replication is the encapsidation of the pgRNA by core protein, forming an immature nucleocapsid. This process requires the cis-acting packaging signal (a stem-loop structure termed as epsilon, ‘ε’) at the 5′ end of pgRNA, and phosphorylation of C-terminal of the core protein [15]. Reverse transcription: tyrosine residue hydroxyl group of polymerase protein covalently binds at the ε region of 5′ pgRNA initiating the reverse transcription [14]. Next, the first three nucleotides from the bulge region of ε stem-loop are synthesized, and the polymerase with the covalently attached trinucleotide sequence translocates from ε to direct repeat, DR1 located at the 3′-end of the 3.5-kb pregenomic RNA. Following the minus-strand elongation, pgRNA template is degraded by an RNase H encoded within the pol protein [28]. Pus-strand DNA synthesis: the terminal 16–18 nts from the 5′ end of pgRNA remains uncleaved and serves as the primer for plus-strand DNA synthesis after it translocates to a complementary sequence on the minus-strand template [29]. Circularization then occurs, facilitated by the short terminal redundancy on the minus strand. Elongation and completion of plus-strand DNA synthesis yield a relaxed-circular DNA genome [14]. Virions secretion: HBV genome is packaged into an icosahedral capsid composed of the HBV core protein (HBc), termed nucleocapsid. RC DNA-containing mature nucleocapsids are enveloped by HBV envelope glycoproteins proteins through the ESCRT machinery in the Golgi and secreted extracellularly as complete virions. A portion of capsids are transported back to the nucleus to maintain the pool of cccDNA[30]. cccDNA, covalently closed circular DNA; pgRNA, pregenomic RNA; RC DNA, relaxed circular DNA; HBeAg,hepatitis B e antigen; HBsAg, hepatitis B surface antigen; mRNA, messenger RNA; Pol, HBV polymerase ; Core, HBV core protein.
2.1. HBV cccDNA
The replication cycle of HBV (Figure 1) begins with the reversible binding of the small envelope protein (S-HBsAg) to heparan sulfate proteoglycans (HSPG) on the hepatocyte membrane (Figure 1)[19]. Recently, Verrier et al. [17] demonstrated that glypican-5 (GPC5), a protein associated with proteoglycans, functions as an attachment factor during the entry of HBV. Following the proteoglycan binding, a specific HBV entrance receptor, Sodium Taurocholate Cotransporting Peptide (NTCP), is recognized by the preS1 region of HBV large envelope protein (L-HBsAg) and it is the N-terminal myristoylated peptide corresponding to amino acids (aa) 2–48 of the pre-S1 that binds to NTCP with high affinity [18]. Upon entrance, capsids are transported within the cytosol to the nucleus during which uncoating and release of the viral genome is initiated. Once the rcDNA enters the nucleus, it undergoes processing to be converted into viral replication intermediates, (covalently closed circular DNA, cccDNA), which is the template for transcription of all viral RNAs [14,15][14][15]. The cccDNA is a stable nuclear form of the viral genome representing the molecular persistent reservoir of HBV. The cccDNA exists in the hepatocyte nuclei as a minichromosome that can be modified by host histone proteins (H3, H4) and non-histone viral and cellular proteins (HBx, HBc, host epigenetics-related proteins). The cccDNA pool is highly stable in hepatocyte nuclei because cccDNA structure is hard to eliminate, and infection of new hepatocytes by progeny virions can be continuous [32,33][31][32]. Although HBV cccDNA lifetime is still unknown, it can persist throughout the natural lifespan of hepatocytes. Therefore, reactivation of viral replication from persistent cccDNA is the principal source of recurrence of clinical hepatitis in patients after stopping antiviral therapy, or after being immunocompromised due to chemotherapy, organ transplantation, immunosuppressive therapy, or coinfection with HIV. It has been established that only a few cccDNA copies per liver can reactivate full virus production. Hence, any cure of CHB requires the elimination of cccDNA or, at least, permanent silence to achieve a functional cure that is sustained undetectable HBV surface antigen (HBsAg) and HBV DNA levels in serum, with or without the appearance of antibodies to the HBsAg (anti-HBs) [28,34][28][33].
Although many aspects of the HBV replication cycle are well characterized, the molecular basis of rcDNA to cccDNA conversion remains unclear, except that the multiple complex steps are necessary. Formation of cccDNA requires multiple enzymatic reactions for the release of the viral polymerase covalently attached to the minus DNA strand, the removal of the RNA primer from the plus DNA strand, the competition of positive-strand, as well as the ligation of the two viral DNA strands [28]. Considering the small viral genome, all the activities, or at least most probably come from the host cellular DNA damage response. In 2014, tyrosyl DNA phosphodiesterase-2 (TDP2) was identified as the first host DNA-repair factor involved in the formation of HBV cccDNA. Recent studies have demonstrated that TDP2 can release the RT from the 5′ end of minus-strand DNA in vitro [26]. A few years later, the flap endonuclease 1 (FEN1) has been reported to remove the 5′-flap structures formed by RNA oligomer and the redundant fragment (r sequence) [27]. Following the removal of flap structures in rcDNA, the plus-strand DNA must be next extended by HBV polymerase and few host cellular polymerases. DNA polymerase κ (POLK) was reported as a key cellular factor involved in cccDNA formation, which supports HBV infection. However, DNA polymerase L (POLL) and H (POLH) were also demonstrated to participate in this process [28]. Recent studies have revealed that both linear strands are ligated by host DNA ligases: LIG I and LIG III. Additionally, LIG IV was also considered to play a role in forming defective cccDNA from the double-stranded linear (dslDNA) through nonhomologous end joining (NHEJ) DNA repair pathway [29,30][29][30].
Despite huge progress in the study of cccDNA that has been made recently, there are many unsolved issues left which might be crucial for developing strategies for a cure of chronic hepatitis B. Thus, further basic studies need to determine other host factors involved in cccDNA synthesis which could help in a deeper understanding of cccDNA formation mechanisms. Moreover, since the amount and transcriptional activity of cccDNA is important for evaluating disease activity and treatment response, it is urgently important to identify novel serum markers that could reflect intrahepatic cccDNA contents and activity.
References
- Nguyen, M.H.; Wong, G.; Gane, E.; Kao, J.H.; Dusheiko, G. Hepatitis B Virus: Advances in Prevention, Diagnosis, and Therapy. Clin. Microbiol. Rev. 2020, 33, doi:10.1128/CMR.00046-19.
- WHO. Global Hepatitis Report, 2017; WHO: Geneva, Switzerland, 2017.
- Chinese Society of Infectious Diseases, Chinese Medical Association; Chinese Society of Hepatology, Chinese Medical Association. The guidelines of prevention and treatment for chronic hepatitis B (2019 version). Zhonghua Gan Zang Bing Za Zhi 2019, 27, 938–961, doi:10.3760/cma.j.issn.1007-3418.2019.12.007.
- Vlachogiannakos, J.; Papatheodoridis, G.V. New epidemiology of hepatitis delta. Liver Int. 2020, 40, 48–53, doi:10.1111/liv.14357.
- Zoulim, F.; Durantel, D. Antiviral therapies and prospects for a cure of chronic hepatitis B. Cold Spring Harb. Perspect. Med. 2015, 5, doi:10.1101/cshperspect.a021501.
- Stasi, C.; Silvestri, C.; Voller, F. Emerging Trends in Epidemiology of Hepatitis B Virus Infection. J. Clin. Transl. Hepatol. 2017, 5, 272–276, doi:10.14218/jcth.2017.00010.
- Joshi, S.S.; Coffin, C.S. Hepatitis B and Pregnancy: Virologic and Immunologic Characteristics. Hepatol. Commun. 2020, 4, 157–171, doi:10.1002/hep4.1460.
- Spearman, C.W.; Afihene, M.; Ally, R.; Apica, B.; Awuku, Y.; Cunha, L.; Dusheiko, G.; Gogela, N.; Kassianides, C.; Kew, M.; et al. Hepatitis B in sub-Saharan Africa: Strategies to achieve the 2030 elimination targets. Lancet Gastroenterol. Hepatol. 2017, 2, 900, doi:10.1016/S2468-1253(17)30295-9.
- Pinho-Nascimento, C.A.; Bratschi, M.W.; Höfer, R.; Soares, C.C.; Warryn, L.; Pečerska, J.; Minyem, J.C.; Paixão, I.C.N.P.; Baroni de Moraes, M.T.; Um Boock, A.; et al. Transmission of Hepatitis B and D Viruses in an African Rural Community. mSystems 2018, 3, doi:10.1128/msystems.00120-18.
- Shan, S.; Cui, F.; Jia, J. How to control highly endemic hepatitis B in Asia. Liver Int. 2018, 38, 122–125, doi:10.1111/liv.13625.
- Walayat, S.; Ahmed, Z.; Martin, D.; Puli, S.; Cashman, M.; Dhillon, S. Recent advances in vaccination of non-responders to standard dose hepatitis B virus vaccine. World J. Hepatol. 2015, 7, 2503–2509, doi:10.4254/wjh.v7.i24.2503.
- Das, S.; Ramakrishnan, K.; Behera, S.K.; Ganesapandian, M.; Xavier, A.S.; Selvarajan, S. Hepatitis B Vaccine and Immunoglobulin: Key Concepts. J. Clin. Transl. Hepatol. 2019, 7, 165–171, doi:10.14218/JCTH.2018.00037.
- Catton, M.; Gray, G.; Griffin, D.; Hasegawa, H.; Kent, S.J.; Mackenzie, J.; McSweegan, E.; Mercer, N.; Wang, L. 2017 international meeting of the Global Virus Network. Antiviral Res. 2018, 153, 60–69, doi:10.1016/j.antiviral.2018.02.001.
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686, doi:10.1016/j.virol.2015.02.031.
- Wang, J.; Huang, H.; Liu, Y.; Chen, R.; Yan, Y.; Shi, S.; Xi, J.; Zou, J.; Yu, G.; Feng, X.; et al. HBV Genome and Life Cycle. In Hepatitis B Virus Infection; Springer: Singapore, 2020; Volume 1179, pp. 17–37, ISBN 1904933203.
- Lazarevic, I.; Banko, A.; Miljanovic, D.; Cupic, M. Immune-escape hepatitis B virus mutations associated with viral reactivation upon immunosuppression. Viruses 2019, 11, 778, doi:10.3390/v11090778.
- Sureau, C.; Salisse, J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology 2013, 57, 985–994, doi:10.1002/hep.26125.
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoudé, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016, 63, 35–48, doi:10.1002/hep.28013.
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049, doi:10.7554/eLife.00049.
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724, doi:10.1073/pnas.1518090112.
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241, doi:10.1016/j.jhep.2018.08.012.
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984, doi:10.1136/gutjnl-2015-309809.
- Schinazi, R.F.; Ehteshami, M.; Bassit, L.; Asselah, T. Towards HBV curative therapies. Liver Int. 2018, 38, 102–114, doi:10.1111/liv.13656.
- Königer, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253, doi:10.1073/pnas.1409986111.
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog. 2018, 14, e1007124, doi:10.1371/journal.ppat.1007124.
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase κ Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893, doi:10.1371/journal.ppat.1005893.
- Long, Q.; Yan, R.; Hu, J.; Cai, D.; Mitra, B.; Kim, E.S.; Marchetti, A.; Zhang, H.; Wang, S.; Liu, Y.; et al. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog. 2017, 13, e1006784, doi:10.1371/journal.ppat.1006784.
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506, doi:10.1038/nrm.2017.48.
- Wang, J.; Shen, T.; Huang, X.; Kumar, G.R.; Chen, X.; Zeng, Z.; Zhang, R.; Chen, R.; Li, T.; Zhang, T.; et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J. Hepatol. 2016, 65, 700–710, doi:10.1016/j.jhep.2016.05.029.
- van Campenhout, M.J.H.; van Bömmel, F.; Pfefferkorn, M.; Fischer, J.; Deichsel, D.; Boonstra, A.; van Vuuren, A.J.; Berg, T.; Hansen, B.E.; Janssen, H.L.A. Host and viral factors associated with serum hepatitis B virus RNA levels among patients in need for treatment. Hepatology 2018, 68, 839–847, doi:10.1002/hep.29872.
- van Bömmel, F.; Bartens, A.; Mysickova, A.; Hofmann, J.; Krüger, D.H.; Berg, T.; Edelmann, A. Serum hepatitis B virus RNA levels as an early predictor of hepatitis B envelope antigen seroconversion during treatment with polymerase inhibitors. Hepatology 2015, 61, 66–76, doi:10.1002/hep.27381.
- Jansen, L.; Kootstra, N.A.; van Dort, K.A.; Takkenberg, R.B.; Reesink, H.W.; Zaaijer, H.L. Hepatitis B Virus Pregenomic RNA Is Present in Virions in Plasma and Is Associated With a Response to Pegylated Interferon Alfa-2a and Nucleos(t)ide Analogues. J. Infect. Dis. 2016, 213, 224–232, doi:10.1093/infdis/jiv397.
- Huang, Y.-W.; Takahashi, S.; Tsuge, M.; Chen, C.-L.; Wang, T.-C.; Abe, H.; Hu, J.-T.; Chen, D.-S.; Yang, S.-S.; Chayama, K.; et al. On-treatment low serum HBV RNA level predicts initial virological response in chronic hepatitis B patients receiving nucleoside analogue therapy. Antivir. Ther. 2015, 20, 369–375, doi:10.3851/IMP2777.