Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Linda Sukmarini and Version 2 by Rita Xu.
The marine environment presents a favorable avenue for potential therapeutic agents as a reservoir of new bioactive natural products. Due to their numerous potential pharmacological effects, marine-derived natural products—particularly marine peptides—have gained considerable attention. These peptides have shown a broad spectrum of biological functions, such as antimicrobial, antiviral, cytotoxic, immunomodulatory, and analgesic effects. The emergence of new virus strains and viral resistance leads to continuing efforts to develop more effective antiviral drugs.
- antiviral peptides
- infectious diseases
- marine peptides
1. Introduction
Infectious diseases, mainly caused by viral pathogens, remain a primary global health issue that has already caused a high mortality rate [1]. Many of the most threatening or deadliest human infectious diseases are caused by viruses, such as human immunodeficiency viruses (HIVs) [2][3][2,3], influenza viruses [4][5][4,5], hepatitis viruses [3][6][3,6], and the recent emerging pandemic threat of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [7][8][7,8]. In spite of striking development in vaccines and small-molecule antiviral drugs, new emerging and re-emerging viral disease outbreaks and the progression of antiviral drug resistance and drug toxicity have forced scientists to perpetually probe novel antiviral agents. Moreover, only a few therapeutic drugs are available for various viruses. Due to their excellent efficacy, high selectivity, and low potential for resistance development, the development of peptide-derived drugs to overcome their constraints on stability and bioavailability is becoming a point of interest in the pharmaceutical industry [9][10][11][9,10,11]. Hence, antiviral peptides (AVPs) that mainly originate from antimicrobial peptides (AMPs) with antiviral activities can be prospective antiviral agents to fight viral infections.
AVPs are typically short peptides (generally consisting of 12–50 amino acid residues) with positively charged (typically +2 to +9) and amphipathic structures [12][13][14][15][16][17][12,13,14,15,16,17]. In addition to these general features enabling these peptides to serve as antimicrobials (including antibacterials, antifungals, and antiparasitics), hydrophobicity is likely to be a key characteristic for AVPs to target enveloped viruses [18][19][18,19]. It is worth mentioning that previous statistical-analysis-based studies on the Antimicrobial Peptide Database (APD) have shown that hydrophobic cysteine residues are abundant in AVPs [20][21][20,21].
Interestingly, as a part of the immune system in all living organisms, these promiscuous peptides are the first line of defense against various pathogens, including viruses. Naturally occurring AMPs with antiviral properties have been found in almost all multicellular organisms, ranging from plants, animals, mammals, and microbes to marine entities. Marine organisms are highly regarded reservoirs of pharmacologically active molecules, including peptides. These natural-product-based peptides evolve naturally through structural modification to adapt to a harsh marine environment. The adapted features eventually enable them to sustain biological properties against pathogens; thus, marine-based peptides have been continuously considered as potential anti-infective drug candidates [22][23][22,23].
2. Biosynthesis of AVPs: A Brief Overview
Biosynthetically generated through ribosomal or non-ribosomal machinery, the vast chemical structures of peptides from natural sources—including AVPs—can differ from linear to cyclic, incorporating canonical and non-canonical amino acids. Essentially, the ribosomal peptides, also designated as ribosomally synthesized and post-translationally modified peptides (RiPPs), are built upon a set of only 20 standard canonical amino acid residues, while non-ribosomal peptides (NRPs) can also contain a larger pool of building blocks of both canonical and non-canonical amino acids [24][25][34,35]. The chemical diversity of RiPPs is generated by the extensive post-translational modifications (PTMs) whereby their precursor peptides are modified by dedicated modifying enzymes encoded in the biosynthetic gene cluster. In brief, the modified peptides subsequently undergo proteolytic cleavage of the leader peptide sequences in the precursor peptides, and additional PTMs of further modified peptides can be present in certain conditions. Thus, this leads to the export of mature and active peptides from the cells [25][26][35,36]. On the other hand, being decoupled from the ribosome, NRPs are synthesized by a multifunctional modular enzyme complex, namely, non-ribosomal peptide synthetases (NRPSs). This machinery assembly line generally consists of initiation, elongation, and termination modules. Each module comprises at least three catalytic domains: (1) an adenylation domain to select a specific amino acid monomer, (2) a thiolation domain to covalently bind the activated monomer, and (3) a condensation domain to catalyze peptide elongation. The product release of NRPS assembly termination can be linear, cyclic, or cyclodepsipeptides. Moreover, further extensive explanations of the NRPs’ synthesis have been reviewed so far [26][27][28][36,37,38].3. Antiviral Mechanism of Action of AVPs
As specific antiviral drugs are mainly dedicated to treating particular viruses, different phases of the viral life cycle have been used to search for novel antiviral drugs. AVPs act against enveloped viruses by interrupting the fundamental stages of their life cycle of entry, synthesis, or assembly. Their inhibition sites include the viral particle or virion inhibition (virucidal effect), adsorption (cellular association), viral penetration, endosomal escape, viral uncoating, viral genome replication, and viral assembly, packaging, and release [14][29][14,39]. Moreover, the proposed mechanism of action of antiviral activity of AVPs may generally encompass (1) their direct binding to the viral target, which is involved in the direct inhibition of host cell infection or viral pathogenesis; (2) their attachment to the target on the host surface (indirect inhibition), which is engaged in the competition or interaction with functional surface proteins of viruses; and (3) their indirect virus-targeting function through suppression of the viral gene expression, as well as inhibition of the viral enzymes, e.g., viral polymerase and integrase, related to the intracellular replication and transcription (biological function inhibition) (Figure 1) [29][30][31][32][33][29,39,40,41,42].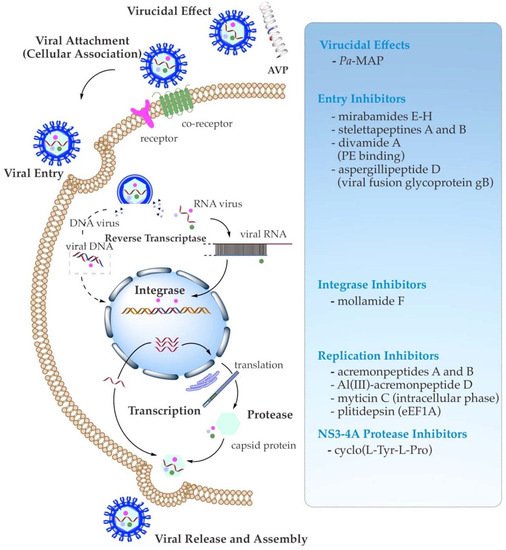
Figure 1. Schematic summary of known antiviral mechanisms of the recently reported marine AVPs.
3.1. Direct Binding Inhibition (Virucidal Effect)
The binding to host cells through interaction with functional receptors is the initial trend in viral infection. Once the viral particle attaches to a host cell, its genetic material is inserted into the cell during this initial attachment and penetration stage. Regarding direct virion inhibition, the AVP papuamide A has shown an immediate virucidal effect of HIV-1 inhibition through a viral-membrane-targeting mechanism [34][35][43,44]. This marine peptide bears a tyrosine hydroxy and an aliphatic moiety tail that have been suggested to interact with the cholesterol membrane of the virus target and assist with insertion into the viral membrane, respectively, resulting in the subsequent viral membrane disruption and, ultimately, viral inactivation [35][44].3.2. Viral Attachment (Cellular Association) and Entry Inhibition
The indirect binding, as evidenced by the HIV-1-targeting α-defensin human neutrophil peptide HNP-1, has been found to bind viral envelope Env and host CD4 glycoproteins/co-receptors in a glycan- and serum-independent manner. In addition, the oligomerization or refolding of HNP-1 could block the viral fusion. Hence, this peptide requires a multifaceted mode of action for HIV entry inhibition [36][37][45,46]. Moreover, several studies [32][38][39][40][41][41,47,48,49,50] have reported that two mechanisms of the cellular association or viral attachment may prevent the entry of the enveloped viruses. In the instance of the activity towards the influenza virus, firstly, the AVP competes with sialic acid to bind with the envelope glycoprotein—namely, hemagglutinin (HA)—and clogs its receptor site, inhibiting the influenza virion from interacting with the host cell membrane. The peptide can imitate sialic acid’s behavior to be recognized by the receptor-binding site of viral HA. Furthermore, the binding of AVP to the other main component of the influenza virus—namely, N-acetylneuraminic acid—on the host cell surface can also prevent viral attachment to host cells. Secondly, the conformation of HA is inhibited; thus, intracellular entry is prohibited, leading to endosomal escape and viral genome release [32][41]. A similar antiviral activity targeting primary attachment has also been found to inhibit HSV and hepatitis viruses. The AVP with a positive charge and good hydrophobicity binds to a cellular glycan moiety to prevent HSV from attaching to the host cell surface [42][51]. This glycan moiety is heparan sulfate, which is known to be a negatively charged glycosaminoglycan that favors HSV viral particles through its basic positively charged binding pocket in the virion glycoprotein to attach to the host cell surface. Therefore, the AVP can prevent HSV virion invasion by binding to glycosaminoglycan molecules as a receptor, hampering the interaction of the virus and the receptor in a host cell. With respect to hepatitis viruses—particularly HCV—some AVPs have also been demonstrated to interact with the virion receptors and co-receptors. AVPs that resemble the cellular protein apolipoprotein E (ApoE) can break up the glycan-dependent interaction or attachment of HCV, hindering entry and infection of target host cells (hepatocytes) [43][44][52,53]. As observed by Chi et al. (2016) [45][54], the HCV fusion-inhibitory peptide could block viral envelope glycoproteins’ E1/E2-mediated membrane fusion by interfering with E1 and E2 heterodimerization. Moreover, the peptide was likely to provoke the dimer E1/E2 glycoproteins’ conformational changes, impairing HCV membrane fusion. Additionally, another AVP (CL58) seemed to inhibit viral entry, possibly after initial binding (post-binding) of the co-receptor cellular membrane protein CD81, and just prior to the final intracellular fusion in endosomes [46][47][55,56]. Furthermore, these mentioned inhibition mechanisms of the viral membrane fusion stage by peptides have been further comprehensively discussed elsewhere [48][57].3.3. Viral Enzymes and Replication Inhibition
In addition, AVPs have also been reported to inhibit the viral replication of HSV-2, influenza viruses, and HCV. In the case of HSV-2, the AVPs can block the transport of a primary viral protein named VP16 into the nucleus. This transcriptional protein regulator induces immediate expression of viral genes required for survival at the initial cellular response. Some AVPs that act on influenza viruses also target viral-RNA-dependent RNA polymerase, comprising PB1, PB2, and PA subunits. These subunits control polymerization and endonuclease cleavage, recognition, binding to the host mRNAs, and endonuclease activity towards host pre-mRNA. Binding those subunits, AVPs can also prohibit the assembly of the influenza polymerase complex [13][32][49][13,41,58]. For HCV, AVPs can act on NS3-4A—a multifunctional enzyme with serine proteinase and helicase functions that are harnessed for HCV replication [50][51][59,60]. Furthermore, to inhibit the replication of the very recent virus SARS-CoV-2, Tonk et al. (2021) [52][61] and Heydari et al. (2021) [53][62] gathered findings on a number of antiviral activities of AVPs that could be involved. Some AVPs directly act on the viral envelope (virucidal effect), binding to the viral spike glycoprotein that blocks the interaction with angiotensin-converting enzyme-2 (ACE2) in host cells, hampering endosomal acidification for uncoating throughout the initial viral life cycle, or escorting the host ACE2 receptor [54][55][56][57][58][63,64,65,66,67]. Moreover, several approaches have been deployed to evaluate the antiviral activity of marine peptides, including neutralization, viral titration, cell viability, and virus plaque-reduction assays [59][27]. Some selected examples of marine AVPs that possess antiviral activity against important human enveloped viruses—such as HIV, influenza, HSV, HCV, and even SARS-CoV-2—are summarized in Figure 1, listed in Table 1, and featured below.Table 1. Recent reported antiviral bioactive peptides derived from marine organisms during 2011–2021.
| Targeted Virus | Peptide | Biosynthetic Class | Origin | IC | 50 | /EC | 50 | /SI/Infectivity | Mechanism of Antiviral Action (Target of Inhibition) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| HIV-1 | Mirabamides E–H | Cyclodepsipeptides/NRPs | Sponge | Stelletta clavosa | 121, 62, 68, 41 nM | Viral fusion | [60] | [68] | ||
| HIV-1 | Stellettapeptines A and B | Cyclodepsipeptides/NRPs | Sponge | Stelletta | sp. | 23 and 27 nM | Viral entry (Viral membrane) |
[61] | [69] | |
| HIV-1 | Mollamide F | Cyclodepsipeptide/NRP | Tunicate | Didemnum molle | PNG07-2-050 |
78 μM (cytoprotective) 39 μM (HIV-integrase) | Viral integrase | [62] | [70] | |
| HIV-1 | Malformin C | Cyclopeptide/NRP | Endophytic fungus | Aspergillus niger | SCSIO Jscw6F30 |
1.4 μM | ND * | [63] | [71] | |
| HIV-1 | Divamide A | Lanthipeptide/ribosomal peptide | Tunicate | Didemnum molle | E11-036 | 0.225 μM | PE binding | [64] | [72] | |
| H1N1/H3N2 | Asperterrestide A | Cyclopeptide/NRP | Endophytic fungus | Aspergillus terreus | 20.2 and 0.41 μM | ND * | [65] | [73] | ||
| HSV-1 | Aspergillipeptide D | Cyclopeptide/NRP | Endophytic fungus | Aspergillus | sp. SCSIO 41501 | 9.5 μM (HSV-1) 12.5 M (ACV-HSV-1) |
Viral intercellular spread (Viral glycoprotein gB) |
[66] | [74] | |
| HSV-1 | Aspergillipeptide E | Linear peptide/NRP | Endophytic fungus | Aspergillus | sp. SCSIO 41501 | 19.8 μM | ND * | [66] | [74] | |
| HSV-1 | Simplicilliumtide J | Cyclodepsipeptide/NRP | Fungus | Simplicillium obclavatum | EIODSF 0210 | 14.1 μM | ND * | [67] | [75] | |
| HSV-1 | Verlamelines A and B | Cyclodepsipeptide/NRPs | Fungus | Simplicillium obclavatum | EIODSF 0210 | 16.7 and 15.6 μM | ND * | [67] | [75] | |
| HSV-1 | Acremonpeptides A and B | Cyclopeptide/NRPs | Fungus | Acremonium persicinum | SCSIO 115 | 16 and 8.7 μM | Viral replication | [68] | [76] | |
| Al(III)-acremonpeptide D | Cyclopeptide/NRPs | Fungus | Acremonium persicinum | SCSIO 115 | 14 μM | Viral replication | [68] | [76] | ||
| HSV-1/HSV-2 | Myticin C | Ribosomal peptide | Mollusk | Mytilus galloprovincialis | 7.69–8.21/8.32–10.5 | The intracellular phase of viral replication | [69] | [77] | ||
| HSV-1/HSV-2 | Pa | -MAP | Ribosomal peptide | polar fish | Pleuronectes americanus | 82% (45 μM)/90% (23 μM) | Virucidal effect | [70] | [78] | |
| HCV | Cyclo(l-Tyr-l-Pro) diketopiperazine |
Cyclopeptide diketopiperazine/NRP | Endophytic fungus | Aspergillus versicolor | 8.2 μg mL | −1 | NS3-4A protease | [71] | [79] | |
| HCV | Valinomycin; streptodepsipeptides P11A and SV21 |
Cyclodepsipeptides/NRPs | Bacterial symbiont | Streptomyces | sp. SV21 | 0–5% | ND * | [72] | [80] | |
| SARS-CoV-2 | Plitidepsin | Cyclodepsipeptide/NRP | Tunicate | Aplidium albicans | 0.88 nM | Viral replication (eEF1A) |
[72] | [80] |
* ND = not yet described.