B-Cell Lymphoma-extra-large (BCL-xL) is involved in longevity and successful aging,
which indicates a role for BCL-xL in cell survival pathway regulation. Beyond its well described
role as an inhibitor of apoptosis by preventing cytochrome c release, BCL-xL has also been related,
indirectly, to autophagy and senescence pathways. Although in these latter cases, BCL-xL has dual
roles, either activating or inhibiting, depending on the cell type and the specific conditions. Taken
together, all these findings suggest a precise mechanism of action for BCL-xL, able to regulate the
crosstalk between apoptosis, autophagy, and senescence, thus promoting cell survival or cell death.
All three pathways can be both beneficial or detrimental depending on the circumstances. Thus,
targeting BCL-xL would in turn be a “double-edge sword” and therefore, additional studies are
needed to better comprehend this dual and apparently contradictory role of BCL-XL in longevity.
- healthy aging
- apoptosis
- autophagy
- senescence
- longevity
- mitochondria
1. The BCL-2 Protein Family
BCL-2, the founding member of the BCL-2 family of proteins, was first identified while studying the chromosomal translocation t(14;18) which is a hallmark of several human follicular lymphomas[1][2] [1,2]. Nevertheless, BCL-2 function was not elucidated until several years later, when Interleukin 3 (IL-3) dependent mouse cell lines were infected with a retroviral vector to express human BCL-2 protein. In the absence of IL-3, BCL-2 promoted cell survival of the infected cells, although they stopped proliferating[3] [3]. Eventually, more proteins having homology sequences with BCL-2 with the capacity to regulate cell death were found. This set of proteins were labeled as the BCL-2 family of proteins.
Currently, it is known that the BCL-2 gene encodes a 26 kD protein consisting of 329 amino acids with a single highly hydrophobic domain at its C-terminus, which enables it to localize mainly in the outer mitochondrial membrane, and, to a lesser extent, in the nuclear envelope and the endoplasmic reticulum. This BCL-2 family of proteins contains all four BCL-2 Homology (BH) domains (BH1, BH2, BH3, and BH4) and a transmembrane (TM) domain. These BH domains facilitate the family members’ interaction with each other, and can result in a pro- or anti-apoptotic function[4][5][6] [4–6]. More than 600 BCL-2 homologous proteins have been described in metazoan (animal) genomes and are currently available in the BCL2DB database[7] [7]. In order to simplify the study of this large family, members of the BCL-2 family have been classified into three main subgroups by reason of their sequence homology regions[8][9][10] [8–10]. These subfamilies are (i) Anti-apoptotic proteins, (ii) Pro-apoptotic proteins and (iii) BH3-only proteins.
- The anti-apoptotic proteins possess the four BH regions (BH1–4), and include BCL-2, BCL-xL (BCL2-L1), BCL-W (BCL2-L2), A1 (also known as BFL-1 or BCL-2A1), and MCL-1 (Myeloid cell leukemia 1).
- The pro-apoptotic proteins, which include BAX (BCL-2 associated X protein), BAK (BCL-2 antagonist killer), and BOK/MTD (BCL-2-related ovarian killer/Matador), have three BH domains (BH1–3) and are considered the pore-forming executioners.
- BH3-only proteins generally possess one single BH region, the BH3 motif, and are also sometimes considered as a subcategory of pro-apoptotic proteins owing to their pro-cell death nature. Proteins like BID (BH3-interacting domain death antagonist), BIM/BOD (BCL2L11; BCL2-interacting mediator of cell death), BAD (BCL2 antagonist of cell death), PUMA/BBC3 (p53 up-regulated modulator of apoptosis), NOXA (phorbol-12-myristate-13-acetate-induced protein 1), BIK/BLK/NBK (BCL-2 interacting killer), HRK/DP5 and BMF (BCL-2 modifying factor) are included in this subgroup[11]. [11].
The role of the BCL-2 family in the regulation of apoptosis is typically described as the anti-apoptotic and pro-apoptotic BH3-only members existing in a state of competitive flux to influence the activation of the pore forming executioners[12][13] [12,13]. Once the executioners are activated, they form pores in the outer mitochondrial membrane (MOM), triggering outer mitochondrial membrane permeability (MOMP), and subsequently the cell undergoes apoptosis[14][15][16] [14–16].
2. BCL-xL
The human BCL2L1 or BCL-X gene is located in the chromosome 20 (20q11.21). Its DNA contains 58,394 bps (genomic size) and its RNA 2575 bps. The BCL-X gene promoter contains consensus motifs for three transcription factor families, STATs (Signal Transducer and Activator of Transcription), Rel/NF-kB (Rel/Nuclear Factor-κB) and Ets (E26 transformation specific sequence), that have been demonstrated to play an important role in the regulation of the BCL2L1 gene expression[17] [17].
Alternative splicing of the BCL-X gene in human cells results in two major mRNA isoforms: the short isoform BCL-xS (591 bp) that has three exons, and the large isoform BCL-xL (780 bp) that has four exons. Many cis-regulatory elements and trans-acting factors exert combinatorial control of BCL-X splicing. Most known regulators, including Sam68, ASF/SF2 (Alternative Splicing Factor 1 / pre-mRNA-Splicing Factor 2, hnRNPA1 (Heterogenous Nuclear Rinonucleoprotein A1), SRp30c (splicing factor arginine/serine-rich 9 protein), and RBM25 (RNA Binding Motif Protein 25), are able to alter BCL-X alternative splicing in vitro or when they are overexpressed in cell cultures[18] [18].
2.1. BCL-xL Protein Structure
At the protein level, three different transcript variants, which encode distinct isoforms, have been reported. The longer isoform BCL-xL (233 aa) acts as an apoptosis inhibitor and the shorter isoform, BCL-xS (170 aa) acts as an apoptosis activator. The third one, BCL-xβ (227 amino acids) differs from the longer and the shorter isoforms by a modification of the last 45 amino acids, however no specific function has yet been related to this isoform[19] [19]. Figure 1A depicts all three BCL-X isoforms.
Human BCL-xL protein structure is formed by a total of eight α-helices, two of which (α5 and α6) have a central location and are disposed in a parallel fashion[20][21] [20,21]. These central helices contain predominantly hydrophobic residues and are flanked by α3 and α4 on one side and by α1, α2, and α8 on the other side. Helices α1 and α2 are connected by a flexible 60-residue loop, which is characteristic of the BCL-xL proteins. It is indispensable for translocation to the nucleus[22] [22] and is the main site for post-translational modifications (phosphorylation, deamidation, and cleavage), which have been shown to be efficient ways to regulate the anti-apoptotic function of BCL-xL[23] [23]. In the context of the three-dimensional structure of BCL-xL, the BH domains make essential contributions to its tertiary structure: the BH1 and BH2 domains encompass turn regions linking two helices, α4 to α5 (in the case of BH1) and α7 to α8 (in the case of BH2). The BH3 domain is located entirely on α2 whilst the BH4 domain is located on α1 and makes a number of stabilizing hydrophobic contacts with α2, α5, and α6. Recent findings have proven a major structural feature on BCL-xL protein that is a large hydrophobic groove involving the BH1–BH3 domains. This hydrophobic groove represents the region of greatest difference between the pro-survival proteins. In BCL-xL, α3 and α4 are almost parallel and are relatively tightly packed resulting in a more closed groove. Mutagenesis studies confirm that this cleft could be the site of interaction with pro-apoptotic proteins[24] [24]. Figure 1B shows BCL-xL primary, secondary, and tertiary structures.
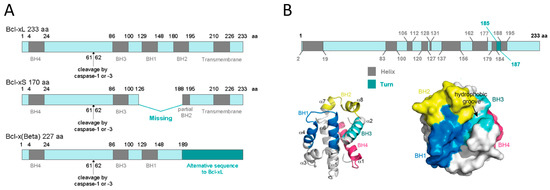
Figure 1. BCL-xL protein structure. (A) BCL-xL, BCL-xS and BCL-xβ isoforms. This representation shows the presence of the BH domains as well as the cleavage site by caspase-1 or -3. (B) BCL-xL primary, secondary, and tertiary structures. The primary structure shows the linear position of helices and turns; the secondary structure represents the 3D position of the eight helices and the four BH domains; and the tertiary structure reveals the hydrophobic groove (adapted from Lee and Fairlie, IJMS 2019[24] [24]).
BCL-xL has dual mechanisms to regulate apoptosis. First, the hydrophobic groove binds to the α-helical BH3 domain of the pro-apoptotic regulators, inhibiting apoptosis. Second, a site distal to the hydrophobic groove binds to cytosolic p53, inhibiting p53-dependent activation of BAX/BAK, and thus, apoptosis[25][26][27] [25–27]. Consistent with the postulated multiple modes of action[28][29][30] [28–30], BCL-xL exists in several conformations, both soluble and in the membrane. Generally, the transition between soluble and membrane conformations can follow two scenarios: anchoring of a relatively unperturbed protein to the lipid bilayer or a complete refolding and bilayer insertion of the protein (e.g., in bacterial toxins). A recent study conducted by Vasquez-Montes et al.[31] [31], demonstrated that the core hydrophobic helix α6 of BCL-xL inserts into the mitochondrial bilayer without adopting a transmembrane orientation. Instead, this insertion disrupts the packing of BCL-xL and releases the regulatory N-terminal BH4 domain (α1) from the rest of the protein structure[31] [31].
At the post-translational level, BCL-xL can be phosphorylated, deamidated, and ubiquitinated. Phosphorylation of BCL-xL at Ser-62 and Ser-49, have been well characterized and can alter BCL-xL intracellular localization and its loop conformation[32] [32], which regulates the molecular association with other proteins such as BH3-only proteins and p53[23] [23]. Interestingly, BCL-xL proteins undergo dynamic phosphorylation/dephosphorylation on Ser-49 and Ser-62 residues during mitosis are important in the maintenance of chromosome integrity in normal cells[33] [33]. The function of BCL-xL is regulated by several kinases, including PLK3 [3[34]4], JNK[35] [35], CDK2[36] [36], and MST1[37] [37]. Deamidation[38] [38] or ubiquitination[39] [39] of BCL-xL are regulated processes that target it for degradation, and can determine susceptibility to DNA-damaging agents and other death stimuli.
BCL-xL Protein Localization
According to The Human Protein Atlas (https://www.proteinatlas.org), at the cellular level, BCL-xL has low RNA cell specificity, as it can be detected in several cell lines such as Caco-2 (colon cancer), MCF7 (mammary gland cancer), HeLa (breast cancer), A549 (lung cancer), etc. In all these cell lines, intracellular localization of BCL-xL protein is mainly in the mitochondria. At the tissue level, BCL-xL has low RNA tissue specificity but the consensus Normalized eXpression (NX) dataset, created as a combination from the three transcriptomics datasets (HPA, GTEx, and FANTOM5), depicts a slight overexpression in bone marrow and thymus. BCL-xL protein levels can be detected in low-medium score in a few tissues, including bone marrow, lymph node, brain, and cells in tubules (Figure 2).
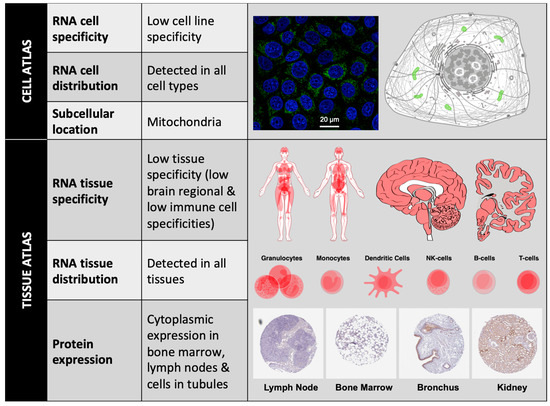
Figure 2. BCL-xL localization. BCL-xL mRNA expression levels can be detected in different cell lines and cell types; BCL-xL protein subcellular location is mainly restricted to mitochondria (Image: MCF7 cells stained with CPTP-BCL2L1-2 antibody obtained from NCI-CPTAC). Consensus data for BCL-xL RNA expression in 55 human tissue types and six blood cell types reveals low tissue specificity. BCL-xL immunohistochemistry in four different tissues stained with HPA035734 antibody (obtained from Atlas Antibodies Sigma-Aldrich). All figures have been obtained from The Human Protein Atlas.