1. Prodrugs Activated by Electron Redistribution
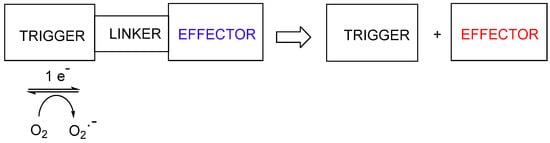
The majority of this class of hypoxia-activated prodrug employs a nitroaromatic trigger unit, suitable because of the unique electronic properties of nitroaromatic compounds, which can accept up to six electrons from various reductase enzymes (e.g., NADPH: cytochrome P450 reductase, ferredoxin: NADP+ reductase), and undergo successive formation of identifiable species: nitro radical anion (A; 1 electron), nitroso (B: 2-electron), hydroxylamine (C; 4-electron) and amine (D; 6-electron) species (Figure 1).
The first one-electron step generates the transient nitro radical anion A, which can be efficiently scavenged (re-oxygenated) in a “futile cycle” by sub-micromolar levels of oxygen
[1][2][6,7], preventing significant further metabolism in well-oxygenated normal tissue. In the hypoxic regions of solid tumours, this re-oxygenation is absent, allowing successive further enzymic reduction of the nitro radical anion to nitroso, hydroxylamine and amine species (B-D) (
Figure 1). This cascade greatly increases the electron density at the nitrogen bearing the substituent R, as shown by the Hammett substituent constants (a measure of the electron-donating or -withdrawing ability of a substituent on an aromatic ring); σ
p NO
2 = +0.78 (electron-withdrawing); σ
p NH
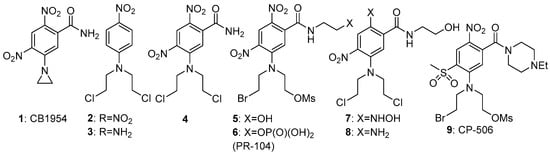
2 = −0.66 (electron-donating). This large change in electron density can be exploited for activation of the group R; for example, greatly enhancing the cellular toxicity of nitrogen mustards as DNA crosslinking agents. An early compound of this general class was the dinitroaziridine CB1954 (
1) (
Figure 2), which was shown
[3][8] to have good bioactivity in rat tumours in vivo, due to activation primarily by the reductase NQO1 (DT-diaphorase). The mammalian version of the enzyme is much less active, limiting the further development of
1, but it remains a useful comparator molecule. A study
[4][9] of substituted
N-phenyl nitrogen mustards showed a difference of up to 7200-fold in toxicity (measured as CT
10 = concentration x time to reduce the surviving fraction to 10% of controls) between compounds
2 and
3 in cultured UV4 CHO cells. While this ratio is augmented by the relatively high sensitivity of UV4 cells, due to defective DNA cross-link removal
[5][10], it nevertheless suggests that adequately high oxic/hypoxic sensitivity ratios can be achieved in vivo.
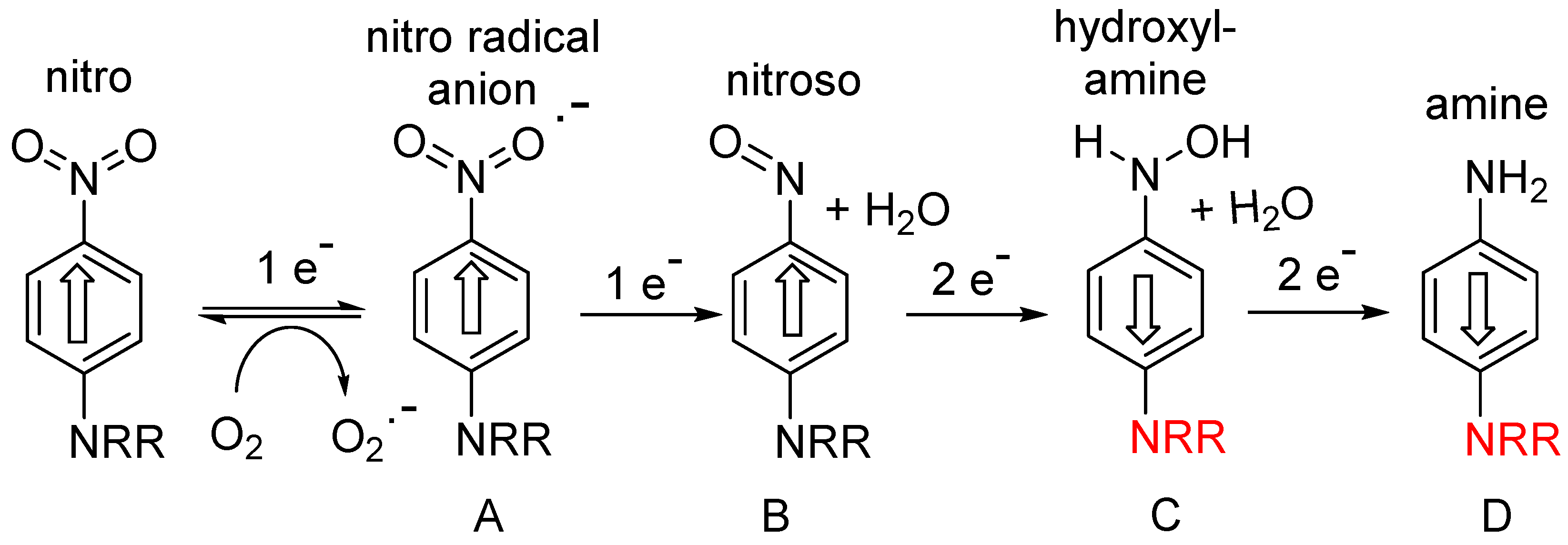
Figure 1. Stepwise reductive activation of nitroaromatic hypoxia-activated prodrugs (after refs.
[6][1][5,6]).
Figure 2. Evolution of PR-104 (6) from CB-1954 (1).
For efficient initial reduction to the nitro radical anion, the reduction potential of the prodrug needs to be within the range of the widespread mammalian reductases noted above (preferably around −350 mV). Despite the effectiveness of
2 in the above UV4 CHO model, with an E(1) of −520 mV it was less effective in a more robust cell model (oxic/hypoxic ratio of about 3-fold)
[4][9]. Increasing the reduction potential of the nitrophenyl mustard
3 by the addition of more electron-withdrawing substituents gave compound
4, with an E(1) of −341 mV and a oxic/hypoxic cell inhibition ratio of 58
[7][11], and this compound was the most effective of a series of regioisomers explored in an SAR (structure-activity relationship) study
[8][12] (
Figure 2).
Further development of this approach
[9][13] using more efficient and more soluble leaving groups on the mustard, resulted in compound
5, which showed improved hypoxic potency (half-maximal inhibitory concentration: IC
50 0.28 µM) and hypoxic/oxic cell ratio (133-fold). The corresponding phosphate (
6), designated PR-104, was taken to Phase II clinal trials by Proacta Inc in 2006, and its major mechanism of action after activation was confirmed to be DNA cross-linking
[10][14]. Hong et al.
[11][15] undertook a detailed study concerning the “bystander effects” of PR-104, using 3-dimensional cell cultures. They concluded that the results could only be explained by the presence of much less polar metabolites than the initial ones, due to exchange of the Br and OSO
2Me units by chloride to give the more rapidly diffusible metabolites
7 and
8. In a mechanistic study
[12][16], the levels of these active metabolites correlated well with the change in DNA reduced crosslinks or monoadducts formed. Increased cytotoxicity of PR-104 was seen in HCT116 cells expressing methionine synthase reductase, novel diflavin oxidoreductase 1 and inducible nitric-oxide synthase, identifying these as effective metabolisers of the prodrug
[13][17]. In a phase I/II clinical trial of PR-104 in patients with relapsed/refractory acute myeloid leukaemia, 32% of patients who received 3 g/m
2 or 4 g/m
2 had a response, with a measurable decrease of the proportions of hypoxic cells present
[14][18]. A spatially resolved pharmacokinetic/pharmacodynamic (SR-PK/PD) model for PR-104, using both single cell suspensions to determine relationships between PR-104A metabolism and clonogenic cell killing, and multicellular layer (MCL) cultures to measure tissue diffusion coefficients, suggested that “bystander effects” contributed 30 and 50% of PR-104 activity in SiHa and HCT116 tumours, respectively
[15][19].
Unpredictable variable toxicity in the Phase II trials stopped further development of PR-104. This toxicity was later shown
[16][20] to be due to aerobic activation by the aldo-keto reductase (AKR1C3) enzyme, not previously recognised as a nitroreductase. Later development of the class resulted in the related compound CP-506 (
9), which did not have this liability and was shown to effectively decrease the hypoxic fraction and inhibit the growth of a wide range of hypoxic xenografted tumours
[17][21]. This compound is currently in Phase II clinical trial
[18][22].
2. Prodrugs Activated by Fragmentation
A second approach to hypoxia-activated prodrugs is the use of a variety of different nitroheterocyclic or nitrophenyl “triggers”, where the electron release on reduction of the nitro group results in fragmentation the linker to release a cytotoxic effector (Figure 3). This approach encompasses a wide variety of cytotoxins of differing mechanisms.
Figure 3. Reductive fragmentation of hypoxia-activated cytotoxins.
This more “modular” design is very versatile, utilising nitrobenzyl, nitrophenyl and a wide variety of nitroheterocyclic trigger units. These have varying reduction potentials and lipophilicity and use effectors encompassing a wide range of different cytotoxic mechanisms when released.
2.1. 4-Nitrobenzyl Triggers
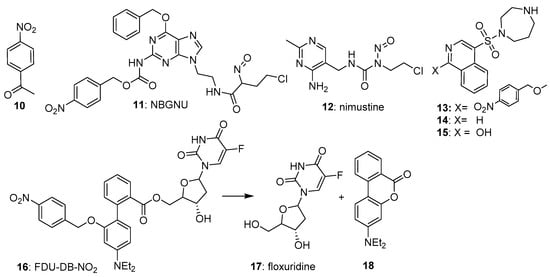
The most widely used nitrobenzyl trigger in prodrugs has been the 4-nitrosubstituted isomer, due partly to its overall higher one-electron reduction potentials (e.g., −356 mV for 4-nitroacetophenone (
10)
[19][23]. Ge et al.
[20][24] reported the 4-nitrobenzyl-triggered hypoxia-activated nitrosourea prodrug NBGNU (
11), which was designed to induce DNA interstrand crosslinks following reduction, by inhibiting O6-alkylguanine-DNA alkyltransferase, an enzyme which has the ability to suppress the chemotherapeutic effect of chloroethylnitrosoureas by removing drug-induced alkyl groups from the O6-site of guanine. In human glioma SF763 cells in culture,
11 was about 5-fold more potent under hypoxic compared to oxic cultures (IC
50s 580 and 126 µM, respectively), with much higher levels of dG-dCd crosslinks in human brain glioma SF126 than cells treated with nimustine (
12) under hypoxia (
Figure 4).
Figure 4. 4-Nitrobenzyl-based prodrugs 10–16.
Al-Hilal et al.
[21][25] reported their design and evaluation of a 4-nitrobenzyl-triggered prodrug (
13) of fasudil (
14), a known inhibitor of the Rho regulator ROCK. Prodrug
13 showed an attenuated IC
50 of 6.8 µM compared with that of fasudil itself (IC
50 0.05 µM) against cells cultured from patients with pulmonary arterial hypertension and grown in air. When the cells were grown under hypoxia,
13 showed an IC
50 of 0.05 µM, undergoing fragmentation to give a mixture of primarily
15 plus some fasudil, with an overall IC
50 of 0.03 µM.
Liu et al.
[22][26] designed a novel prodrug (FDU-DB-NO2:
16) that on bioreductive fragmentation released not only the antimetabolite floxuridine (
17), but simultaneously generated the fluorescent dye 4′-(diethylamino)-1,1′-biphenyl-2-carboxylate (
18); the latter for real-time tracking of drug release. The prodrug
16 was evaluated under normoxic and hypoxic cultures of MCF-7 breast cancer and MCG-803 human gastric cancer cells, where it showed good activity (
Figure 4). In severe hypoxia, the prodrug was highly effective in suppressing ROCK activity in pulmonary arterial smooth muscle and endothelial cells but was less effective in MCF-7 xenografts in nude mice.
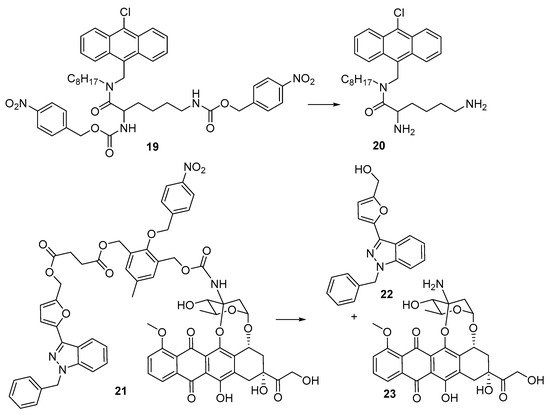
Yeoh et al.
[23][27] reported the preparation and evaluation of the bis(4-nitrophenyl) prodrug (
19) of the anti-bacterial antibacterial peptoid mimic
20 [24][28] against cultures of
S. aureus and
E. coli. The prodrug was inactive with a minimum inhibitory concentration (MIC) >256 µg/mL in both strains, but when reduced (chemically, by SnCl
2) showed MICs of 2–4 µg/mL. Luo et al.
[25][29] developed a dual-release drug (
21), which simultaneously generated YCH-1 hemisuccinate, which rapidly cyclised to give the HIF-1α expression inhibitor YCH-1 (
22) and doxorubicin (
23). In human adenocarcinoma A549 cells the prodrug
21 had an IC
50 of 9.6 μM compared with free doxorubicin (IC
50 4.3 μM) but was considerably more cytotoxic (IC
50 1.2 μM) under hypoxia. Treatment of A549-tumor-bearing mice with doxorubicin (
23) (2.9 mg/kg × 4 daily) or 21 (1.5 mg/kg × 4 daily) showed a 3-fold slower tumour growth with prodrug
21 than with doxorubicin (
Figure 5).
Figure 5. 4-Nitrobenzyl-based prodrugs 19 and 21.
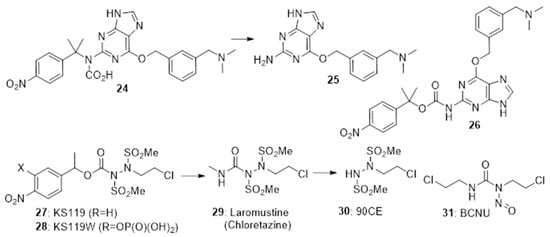
4-Nitrobenzyl triggers have also been used in cases where the chemistry of the rest of the generates fragmentation following nitro-reduction. The prodrug
24 fragments preferentially under hypoxia to release the O6-alkylguanine-DNA alkyltransferase (AGT) inhibitor
25 [26][30]. In EMT6 cells under hypoxia
24 was efficiently converted to
25 and was able to significantly enhance the cytotoxic effect of alkylators. Zhu et al.
[27][31] also prepared the isomeric prodrug
26 which was similarly activated under hypoxia in EMT6 cells >60-fold faster under hypoxic compared to normoxic culture conditions. Noting the effectiveness of chloroethylureas on leukaemia and lymphomas, Sartorelli’s group developed a number of 4-nitrobenzyl prodrugs of this DNA-ethylating agent. The 4-nitrobenzyl prodrugs
27 [28][32] and the more soluble
28 [29][33] both rapidly fragment on nitro group reduction to release the active alkylating agent
29 (chloretazine), which in turn releases the ethylating agent
30. While both clorazetine and the clinical alkylating agent Carmustine (BCNU,
31) both release 2-chloroethyl isocyanate, clorazetine is much more effective than BCNU against human erythrocytes in culture, suggesting that the co-released methyl isocyanate from clorazetine is important
[30][31][34,35] (
Figure 6).
Figure 6. 4-Nitrobenzyl-based prodrugs 24, 26, 27 and 28.
2.2. Nitrophenyl Triggers
These have not been widely used in prodrugs due to their low reduction potential (see the development of PR-104 (
6) above). 4-Nitrobenzoic acid has an E(1) of −425 mV
[1][6], and Francisco da Silva et al.
[32][36] measured reduction potentials of around −1000 mV for a series of 4-nitrophenyl compounds. However, special cases exist. SLC-0111 (
32) is a potent inhibitor of the carbonic anhydrases CAIX and CAXII (particularly of CAXII; Ki 4.4 nM). Nocentinii et al.
[33][37] report the synthesis and evaluation of a number of potential hypoxia-activated prodrugs of
32 (e.g.,
33), using as trigger the 3-nitrophenyl unit in the drug, whose one-electron reduction potential is significantly raised by an attached SO
2NH
2 group. Compound
33 was a reasonably potent inhibitor of the enzyme (Ki of 0.21 µM against CAIX and CAXII) and also showed modest selective potency for hypoxic over oxygenated cultures of MDA-MD-231 adenocarcinoma cell line.
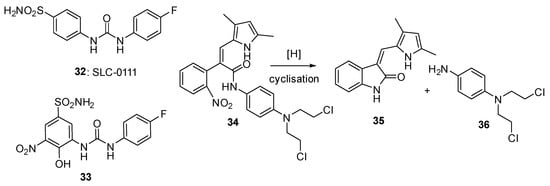
Sansom et al.
[34][38] describe the design and evaluation of an intriguing prodrug (
34) where a 2-nitrobenzyl trigger, when reduced, fragments to simultaneously release the kinase inhibitor semaxanib (
35) together with the nitrogen mustard (
36). The prodrug was evaluated in both aerobic and hypoxic cultures of MDA-MB-468 breast cancer cells, but no hypoxic selectivity was seen. This was likely due to low reduction potentials (measured as −509 mV for prodrug
34) (
Figure 7).
Figure 7. Nitrophenyl-based prodrugs 33 and 34.