Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Shizuka Uchida.
Long non-coding RNAs (lncRNAs) belong to a class of non-protein-coding RNAs with their lengths longer than 200 nucleotides. Most of the mammalian genome is transcribed as RNA, yet only a small percent of the transcribed RNA corresponds to exons of protein-coding genes. Thus, the number of lncRNAs is predicted to be several times higher than that of protein-coding genes. Because of sheer number of lncRNAs, it is often difficult to elucidate the functions of all lncRNAs, especially those arising from their relationship to their binding partners, such as DNA, RNA, and proteins. Due to their binding to other macromolecules, it has become evident that the structures of lncRNAs influence their functions.
- epitranscriptomics
- gene expression
- lncRNA
1. Introduction
By definition, long non-coding RNAs (lncRNAs) are any ncRNAs that are longer than 200 nucleotides (nt). With the advancement of high-throughput techniques [microarrays, next generation sequencing (NGS), especially RNA sequencing (RNA-seq)], many lncRNAs have been discovered [1]. To date, a number of functions of lncRNAs have been proposed and experimentally validated; ranging from decoy, epigenetic, transcriptional, post-transcriptional, and translational controls [2,3,4,5][2][3][4][5]. The general understanding in the field is that lncRNAs exert their actions by binding to other macromolecules, which are DNA, RNA, and proteins [6,7][6][7]. Thus, it is essential to identify the potential binding partners to elucidate the mechanism of action of lncRNAs. To this end, the most popular method is using an affinity tag on an in vitro purified RNA and using this RNA as a bait to pull-down proteins/nucleic acids from cellular extracts. There are other more elaborated methods currently available, including ChIRP (Chromatin isolation by RNA purification), CHART (Capture Hybridization Analysis of RNA Targets), CLIP (cross-linking and immunoprecipitation), and RAP (RNA antisense purification), which are comprehensively reviewed elsewhere [8,9,10][8][9][10].
Just as DNA and proteins, RNA can be modified by a variety of enzymes. The classic example is the RNA modifications of ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs), which affect the efficiency of translation [11,12,13][11][12][13]. To date, there are more than 170 RNA modifications known across species [14], which has opened up a new field of study called, epitranscriptomics [15[15][16],16], whose name is based on the well-studied field of DNA modification, epigenetics. Much of the concepts of epigenetics are applied to dissect the ever-growing field of epitranscriptomics, including the epitranscriptomic enzymes being categorized as writers, readers, and erasers. Among epitranscriptomic marks, the most well studied one in recent years is N6-methyladenosine (m6A), which is a methylation of nitrogen-6 position of adenosine (A) found in messenger RNAs (mRNAs) and non-protein-coding RNAs (ncRNAs). Other epitranscriptomics marks in mammals include the A-to-I RNA editing, 2′-O-methylation (2′-O-Me), N1-methyladenosine (m1A), 3-methylcytidine (m3C), 5-methylcytosine (m5C), N7-methylguanosine (m7G), pseudouridylation (Ψ) to name a few [17,18][17][18]. These epitranscriptomic marks affect all realms of RNA lifecycle, including splicing, subcellular localization, microRNA (miRNA) biogenesis and bindings, RNA stability, and translation efficiency [19,20][19][20]. More importantly, dysregulation of epitranscriptomic marks affect many diseases, including cardiovascular [21], liver [22], and neurodegenerative diseases [18] as well as cancers [23].
2. Epitranscriptomic Marks Affect RNA Structures as in the Case of Immune Responses
RNA exists in a single-stranded (ssRNA) or double-stranded RNA (dsRNA) state. The balance between these states may be influenced by cellular conditions, such as stress and viral infection [25,26,27,28][24][25][26][27]. Furthermore, more than half of the human genome consists of repetitive sequences, such as those derived from transposons and ALU elements [29][28]. These repetitive sequences form palindromic repeats, resulting in the formation of dsRNAs [30][29]. To detect dsRNAs, there are several high-throughput methods available, including PARS (Parallel Analysis of RNA Structure) by sequencing RNA digested with RNases S1 and V1 that specifically recognize single-stranded RNA (ssRNAs) and dsRNAs, respectively [31][30]. Other methods to analyze RNA structures are DMS-Seq to label RNA structures by dimethyl sulfate (DMS) [32][31], LIGR-seq (LIGation of interacting RNA followed by high-throughput sequencing) to globally map RNA–RNA duplexes crosslinked in vivo [33][32], PARIS to detect dsRNA [34][33], RIC-seq (RNA in situ conformation sequencing) to globally profile intra- and intermolecular RNA–RNA interactions [35,36][34][35], SHAPE-Seq (selective 2′-hydroxyl acylation analyzed by primer extension sequencing) [37][36], and SHAPE-MaP (selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling) to chemically probe RNA by adding RNA-specific small molecules in cell culture [38,39][37][38]. Recently, a comprehensive RNA structure probing database, RASP, was released, which contains 18 species (e.g., animals, plants, bacteria, fungi, and viruses) and 18 different experimental methods measuring RNA secondary structures in a transcriptome-wide manner [40][39]. Furthermore, there are databases for epitranscriptomic marks (comprehensively reviewed in [41][40]), including RMBase v2.0 [42][41] and RMVar [43][42] that contain several epitranscriptomic marks for different organisms. It will be of great interest to further analyze the collected data sets by merging them with high-throughput data that map known epitranscriptomic marks. This will enable the analysis of the preferential distribution of each epitranscriptomic mark to ssRNAs and dsRNAs in different species (thus, evolutional-conservation, if any). Upon viral infection, the innate immune system is triggered, which recognizes pathogen-associated molecular patterns (PAMPs, which are unique molecular ligands on or within microbes, including viral DNA and RNA) leading to activation of intracellular signaling pathways to initiate antiviral response [44,45][43][44]. These PAMPs are detected by the host through pattern recognition receptors, such as Nod-like receptors (NLRs), RIG-I-like receptors (RLRs), and Toll-like receptors (TLRs) [46][45]. In the case of RLRs, RIG-I senses short dsRNAs, while the RLR, MDA5 (melanoma differentiation-associated protein 5), detects long dsRNAs. These recognitions of PAMPs by RLRs are followed by MAVS (mitochondrial antiviral-signaling protein)-mediated activation of signaling cascades, including type I interferon responses [47,48,49][46][47][48]. The epitranscriptomic mark, m6A, plays active roles in innate immunity by reducing type I interferon production [50,51][49][50]. Winkler et al. reported that m6A marks deposited by the m6A METTL3 and read by the m6A reader YTHDF2 negatively regulate interferon response by facilitating the fast turnover of interferon mRNAs leading to viral propagation [50][49] (Figure 1A). Interestingly, increasing evidence suggests that lncRNAs are shown to be involved in virus infections and antiviral immune responses [52][51]. Furthermore, many lncRNAs have m6A marks [53[52][53][54],54,55], influencing secondary structures of lncRNAs. For example, MALAT1 (metastasis associated lung adenocarcinoma transcript 1) is involved in inflammatory responses and innate immunity [56,57,58][55][56][57] along with its enzymatic processing product, MALAT1-associated small cytoplasmic RNA (mascRNA) [59,60,61,62,63][58][59][60][61][62]. These findings highlight that further investigation of epitranscriptomic marks on lncRNAs and their secondary structural changes may reveal the active involvement of lncRNAs in innate immunity. In this regard, it will be of high interest to understand the relationship between lncRNAs and another epitranscriptomic mark, pseudouridylation (Ψ) [64][63], as it is demonstrated recently in COVID-19 mRNA vaccines using N1-methylpseudouridine (m1Ψ) to increase their effectiveness [65][64].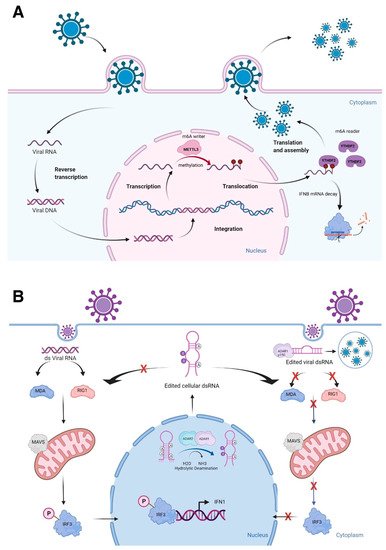
Figure 1. Epitranscriptomic marks and RNA structures in immune responses. (A) Viral RNA methylation deposited by the m6A writer METTL3 and read by the m6A reader YTHDF2 negatively regulate cellular defense response by facilitating the fast turnover of interferon mRNA leading to viral replication. (B) The role of ADARs in differentiating self-from non-self dsRNA by modulating canonical antiviral pathways induced by dsRNA. During an infection, the viral dsRNA enters into the cytoplasm. Non-edited dsRNA binds to MDA5 (melanoma differentiation-associated protein 5) and RIG-I (retinoic acid-inducible gene I like receptor). This complex activates MAVS (mitochondrial antiviral-signaling protein), leading to the phosphorylation of IRF3 (interferon regulatory transcription factor 3) and its translocation into the nucleus, thus inducing a type 1 interferon response. Endogenous cellular dsRNA that is generated during transcription is A-to-I edited by ADARs. The ADAR1 isoform p150 is cytoplasmic and is induced by interferon. It edits dsRNA either of viral or cellular origin. This dsRNA contains inosine and inhibits the activation of MDA5 and RIG-1, thus turning off the interferon response and apoptosis to prevent autoimmune reaction. However, this mechanism could favor virus replication, if it is not tightly regulated. Figure is created with BioRender.com, accessed on 15 March 2022.
A-to-I RNA editing is a type of epitranscriptomic mark that involves the RNA editing enzymes, ADARs [adenosine deaminases acting on RNA, consisting of three genes: ADAR1, ADARB1 (ADAR2), and catalytically inactive ADARB2 (ADAR3)], recognize dsRNAs to catalyze adenosine to inosine (A-to-I) conversion, mostly at ALU repeats and introns [21]. ALU repeats are ~300 bp that belong to the family of repetitive elements in primates. There are more than one million ALU repeats in primate genomes [66][65]. Two transcribed ALU repeats form a quasi-palindrome, which becomes double-stranded RNA to recruit ADARs to catalyze A-to-I RNA editing [67][66]. I is recognized as guanosine (G) by splicing and translational machineries as well as in reverse transcription reactions; allowing detection of A-to-G changes in RNA-seq reads when these reads are mapped to the reference genome [68][67]. Mutations in the human ADAR1 gene result in the autoimmune disease, Aicardi-Goutières syndrome, while the whole-body knockout mice of Adar1 results in embryonic death due to massive apoptosis and aberrant interferon induction, which can be rescued to live birth by ablating the RLRs, Mavs or Mda5 (melanoma differentiation-associated protein 5) [69,70,71][68][69][70]. Both ADAR1 and ADAR2 are important in differentiating self- from non-self dsRNAs [70,72,73][69][71][72] (Figure 1B). Furthermore, silencing of ADAR1 in the human hepatocellular carcinoma cell line, HepG2, resulted in shifting of dsRNAs to ssRNAs at the transcriptome-wide level [74][73]. As many lncRNAs have A-to-I RNA editing sites [75[74][75],76], further characterization of RNA editing sites will uncover the secondary structures of lncRNAs, especially the conversion of A to I at the nitrogen-6 position of adenosine, which can be methylated as m6A, if not edited [77][76]. Thus, both epitranscriptomic marks, A-to-I RNA editing and m6A, could competitively affect the secondary structures of lncRNAs, thereby, influencing the binding of other macromolecules.