Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Amina Yu and Version 4 by Amina Yu.
Carbonic anhydrase (CA, EC 4.2.1.1) IX isoform is a surficial zinc metalloenzyme that is proven to play a central role in regulating intra and extracellular pH, as well as modulating invasion and metastasis processes. With its strong association and distribution in various tumor tissues and well-known druggability, this protein holds great promise as a target to pharmacologically interfere with the tumor microenvironment by using drug combination regimens.
- carbonic anhydrase IX
- small-molecule inhibitors
- solid tumors
- tumor hypoxia
- tumor acidosis combination cancer therapy
- adjuvant agents
1. Acetazolamide in Combination with Conventional Cytostatic Agents
1.1. Acetazolamide and Doxorubicin
Cellular uptake of a wide range of conventional cytostatic agents can be hindered due to the shifts in tumor microenvironment characteristics, such as poor perfusion, hypoxia, and/or acidity [1]. The widely acknowledged ion-trapping model predicts that the decrease in extracellular pH, which is often encountered in hypoxic regions, prevents weekly basic drugs from penetrating malignant cells and thus confers regional failure [1]. Inhibition of CA IX isoform on the cellular surface proved to be a viable approach for affecting extracellular acidification in solid tumors [2]. One of the pioneering attempts to employ small molecular CA IX blockers to overcome cytostatic resistance in hypoxic (acidic) regions was made in 2012 by Geiling and colleagues who investigated the combined effect of CA pan-inhibitor acetazolamide (AZ) and doxorubicin (DOX) [3]. To this end, colon carcinoma HT-29 (CA IX rich) and HCT 116 (CA IX poor) cell lines, as well as MDA-MB-435 human melanoma cells, stably transfected to express empty vector (EV1) or CA IX (CA9/18) (Figure 1). Interestingly, the treatment with AZ significantly increased the toxicity of DOX in CA IX-rich cells. Conversely, DOX efficacy was unaffected by AZ treatment in the cells with low CA IX expression. It is concluded that the effect of AZ treatment is largely related to the blocking of CA IX, which is instrumental for cancer cells to maintain acidic pHe, thus hampering the membrane transport of weakly basic drugs, including DOX. This point was further supported by the fact that the weakly acidic drug melphalan exerted reduced toxicity under the same conditions against the CA IX-rich cell lines (Figure 1) [3].
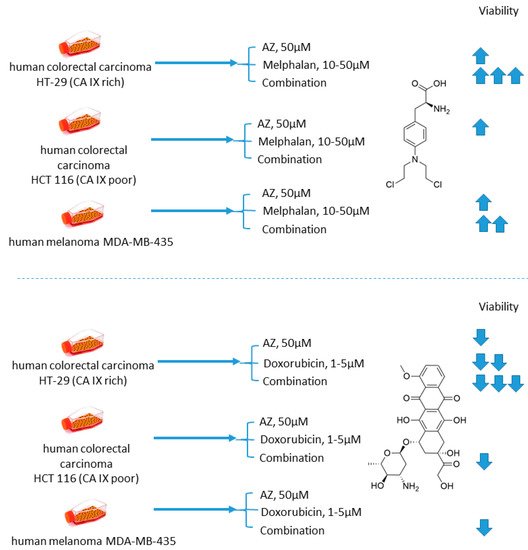
Figure 1. Graphical outline of the study on AZ+melphalan/DOX combination reported by Geiling et al. [4].
1.2. Acetazolamide and Sulforaphane
Mokhtari and coworkers reported a series of studies investigating the synergistic activity of AZ and natural product sulforaphane against bronchial carcinoid cell lines and their derived mice subcutaneous xenografts [54]. Sulforaphane shows multiple anticancer effects [5][6][7][8]. Among others, it selectively activates the nuclear transcription factor erythroid 2p45 (NF-E2)-related factor 2 (Nrf2)-Kelch-like ECH-associated protein 1 (Keap1), which is an essential downstream effector of the PI3K/Akt/mTOR [98][109]. Importantly, overexpression of Nrf2 due to defects in Keap1 was observed in several types of cancer, where it served as a non-HIF mediated mechanism, promoting tumor cell survival in hypoxic conditions, including apoptosis inhibition, metabolic reprogramming, and chemotherapeutic resistance [1110]. Finally, sulforaphane has been reported to reduce the expression of serotonin receptors (5-HT2, 5-HT3) and transporter (SERT) in Caco-2 cells, which appears relevant with regard to the treatment of hormone-secreting bronchial carcinoids [11][12][13].
As unveiled by Mokhtari, combined treatment of NCI-H727 (typical) and NCI-H720 (atypical) lung carcinoid cells with AZ and sulforaphane (SFN) led to a significant reduction in their viability and clonogenicity, as well as markedly decreased the fraction of invasive cells compared to the single-agent controls. In addition, a profound drop in the tumor-derived serotonin has been noticed in vitro. It is suggested that the observed synergistic effects could be explained through the inhibition of both MEK/Erk (MAPK/ERK kinase/ extracellular signal-regulated kinase) and PI3K/Akt (phosphatidylinositol-3 kinase/Akt) pathways, which in turn regulate the expression of CA IX and other HIF-targeted genes [54]. Furthermore, in H727 and H720 spheroids, the AZ+SFN combination largely decreased proliferation rates compared to single-agent therapies. Noticeably, when xenografted into immunocompromised mice, these spheroids still strongly responded to the combined treatment, and a prominent reduction in tumor growth and gross vascularization was observed. Such effect was accompanied by a remarkably decreased expression of the stemness markers. The in vivo studies also further confirmed the downregulation of the PI3K/Akt/mTOR (mechanistic target of rapamycin) pathway and downstream effectors, as well as a more profound perturbation of pH homeostasis in lung cancers under exposure to AZ+SFN treatment. Moreover, these conditions markedly reduced the 5-HT expression in both the atypical H720 and typical H727 bronchial carcinoid xenograft, and the highly aggressive atypical histotype was extremely sensitive to the treatment. Therefore, the combination AZ+SFN promisingly exerted multiple anticancer agents in different models of bronchial carcinoid via blocking a range of hypoxia-mediated pro-survival pathways and 5-HT secretion in bronchial tumors [1413].
The joint action of the drugs against highly aggressive bronchial and bladder cancers was associated with restored pHi and pHe values and a more efficient blockade of PI3K/Akt/mTOR pathway as compared to single-agent treatment. In addition, the combined treatment enhanced apoptosis and produced a significant drop in the expression of adhesive molecules and stemness markers in tumor xenografts, whereas the expression of 5-HT was profoundly affected in the pulmonary carcinoid (Figure 2).
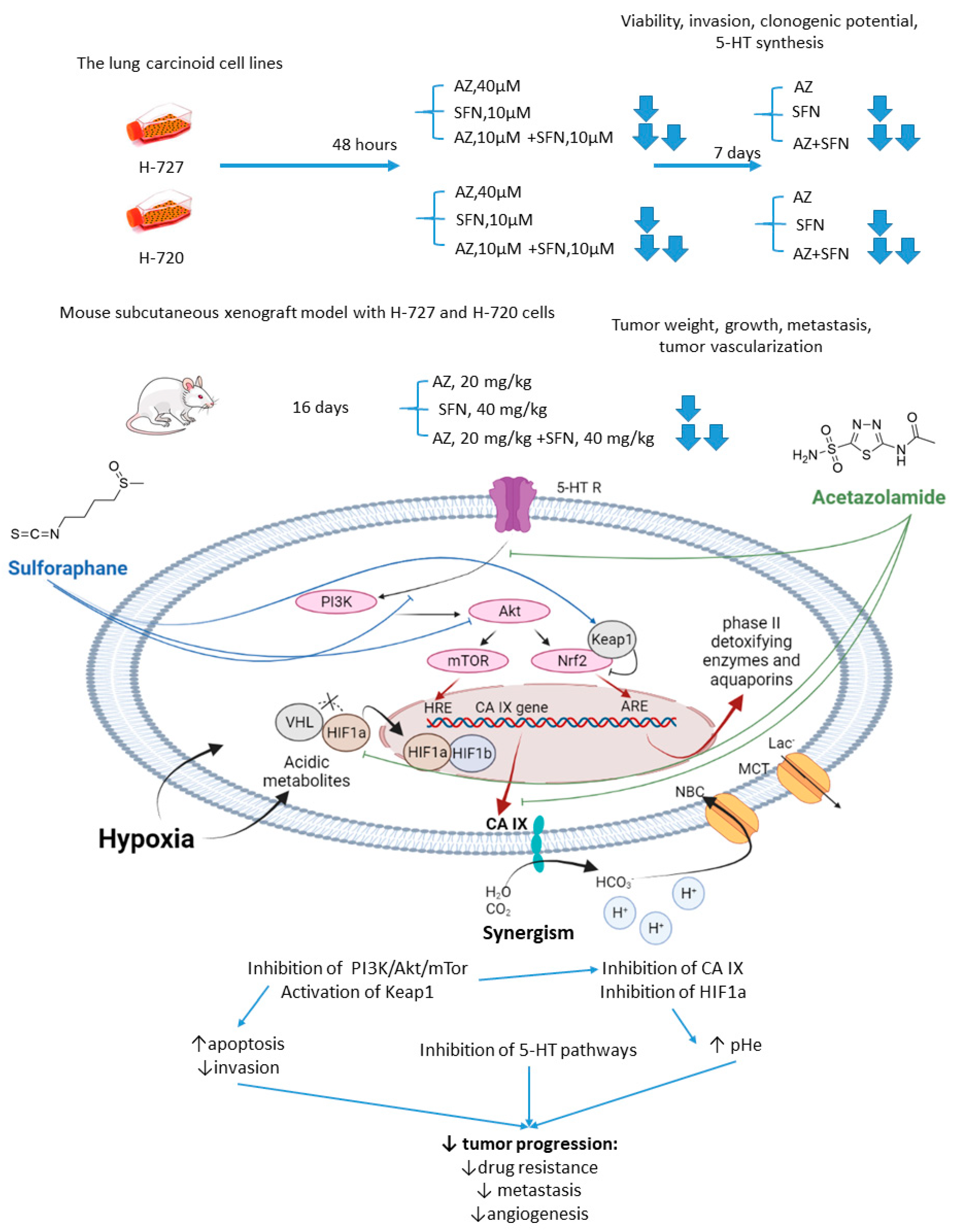
Figure 2. Graphical outline of the study on AZ+SFN combination reported by Mokhtari et al., and summary on the hypothesized molecular interactions resulting in synergistic action of the drugs [15][16].
1.3. Acetazolamide and Cisplatin
Another involving AZ was reported by Gao in 2018 [1714]. AZ was combined with an old DNA-alkylating drug cisplatin (CIS) to treat Hep-2 laryngeal carcinoma cells (Figure 3). The drug combination was more efficient than single-agent treatment and resulted in decreased levels of Hep-2 cell viability, proliferation, and metastasis. Meanwhile, elevated expression of p53 and drop in the Bcl-2/bax ratio corresponded to higher rates of apoptosis in cancer cells. Unexpectedly, in this study, healthy human umbilical vein endothelial cells (HUVEC) turned out to be insensitive to both AZ and CIS either in single-agent or combined regimens, yet there are no evident reasons for such selectivity (at least for cisplatin) (Figure 3) [1714].
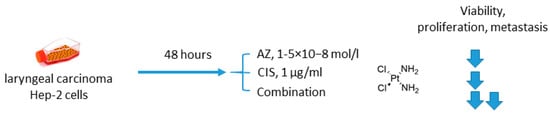
2. Acetazolamide in Combination with Targeted Anticancer Drugs
2.1. Acetazolamide and Rapamycin
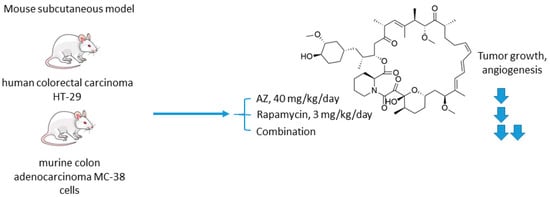
The mechanistic target of rapamycin (mTOR) is a serine/threonine-specific protein kinase that belongs to the family of phosphatidylinositol-3 kinase (PI3K) [1915]. Participating in multiple signaling pathways by forming mTOR complexes 1 and 2 (mTORC1 and mTORC2), this protein regulates cell cycle progression, apoptosis, autophagy, proliferation, and metabolism of tumor cells [2016]. Macrolide’s compound rapamycin and its analogs (also known as rapalogs) represent the first generation mTOR inhibitors [2117]. Despite showing encouraging results in preclinical models as monotherapeutic agents, these drugs demonstrated limited efficacy in patients as tumor relapse often occurred through resistance formation [2218]. In 2016, Faes and coworkers reported that the activity of mTORC1 is mainly restricted to the non-hypoxic tumor compartment, suggesting that there is a potential for combination treatment involving hypoxia-targeting molecules (Figure 4) [2319]. In fact, in vivo experiments on mice injected with colorectal carcinoma HT-29 cells and murine colon adenocarcinoma MC-38 cells highlighted that mTORC1 hyperactivation promotes tumor-cell proliferation in normoxia. Contrastingly, in hypoxic areas of the tumor, a decrease in mTORC1 activity was observed, which abrogated rapamycin antitumor efficacy in these areas. Furthermore, rapamycin treatment increased the hypoxic tumor compartment compared to controls in both HT-29 tumor xenografts and MC-38 tumor allografts and gave significant rise to CA IX protein expression in these regions. Based on these findings, the joint effect of AZ and rapamycin in the mouse models [2319]. It turned out that both AZ and rapamycin alone reduced tumor growth; however, the effect was dramatically increased when both agents were combined. Interestingly, the observed effect was long-lasting, as after three months of the treatment HT-29 tumor xenografts did not progress. It was additionally demonstrated that AZ increased tumor necrosis and the number of tumor blood vessels in HT-29 and MC-38 tumors. Moreover, AZ drastically reduced proliferation in hypoxic, but not in normoxic tumor regions, whereas the opposite was true for rapamycin. Thus, combined administration of rapamycin and AZ produced remarkable antiproliferative effects in vivo in both hypoxic and non-hypoxic compartments (Figure 4) [2319].
2.2. Acetazolamide and Bevacizumab
Vaeteewoottacharn and colleagues investigated AZ as an adjuvant agent for the treatment of cholangiocarcinoma, which has a very poor prognosis and a small range of therapy opportunities [2520]. The overexpression of VEGF in tumor tissue gives rise to the use of VEGF inhibitors for this cancer; however, cancer cells’ adaptation to the treatment resulted in limited efficacy in patients [2621]. Of note, HIF-1α and CA IX upregulation has been reported to contribute to the drug resistance against anti-angiogenic therapy [2722]. In this context, Vaeteewoottacharn combined AZ with bevacizumab, a monoclonal antibody that blocks angiogenesis by inhibiting VEGF to treat cholangiocarcinoma tumors in vivo. Despite the fact that bevacizumab (0.1 mg/kg/mouse) effectively inhibited tumor growth, remarkable overexpression of HIF1α, VEGF, VEGFR1, and CA IX was observed in the treated tumors (Figure 5). The results, therefore, highlighted the compensative mechanism of the tumor in response to the VEGF inhibition. Reassuringly, the combination treatment with AZ produced a more significant reduction in tumor growth and angiogenesis, although the influence of the combined treatment on VEGF and HIF1α pathways remained uninvestigated (Figure 5) [2520].
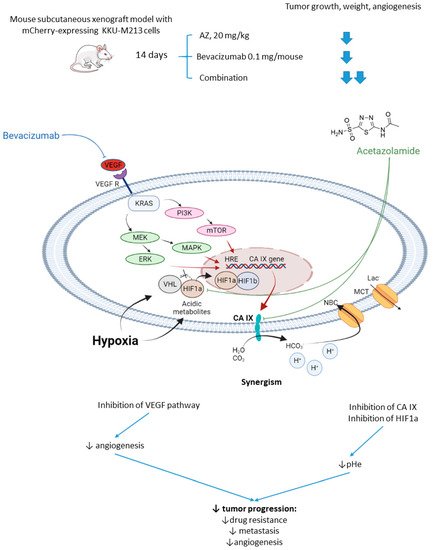
Figure 5. Graphical outline of the study on AZ+bevacizumab combination reported by Vaeteewoottacharn et al. and a summary on the hypothesized molecular interactions resulting in synergistic action of the drugs [28].
2.3. Acetazolamide and Imatinib
In 2017, Abd-El Fattah and colleagues combined imatinib (IM) with AZ in attempts to enhance the cellular uptake of the former weakly basic drug (Figure 6) [2923]. IM(imatinib) is a tyrosine kinase inhibitor capable of blocking tumorigenic and prometastatic kinases Bcr-ABL, c-Kit, platelet-derived growth factor receptor (PDGFR), and epidermal growth factor receptor (EGFR) [3024]. Co-administration of AZ led to a significant increase in the uptake of IM by cells T47D and MCF-7 breast cancer cells. It was reported considerable biochemical alterations in the presence of AZ compared to IM monotherapy. In particular, they observed suppression of HIF-1α mRNA, accompanied by a decrease in VEGF secretion, inhibition of NO release, a profound suppression of matrix metalloproteinases MMP-2 and 9 and phospho-p38 MAPK (mitogen activated protein kinase) and rise in tissue inhibitor of metalloprotease-1, 2 (TIMP-1, 2) levels. These results highlighted the potential of the drug combination in question to block angiogenesis and metastatic potential in solid tumors by affecting multiple signaling pathways. Subsequently, in vivo experiments were performed by using Ehrlich ascites carcinoma (EAC)-bearing mice. It was found that single-agent IM administration caused reduced tumor volume by almost 46% after 3 days of treatment. In the combination regimen, the tumor volume reduction amounted to 63%. Histopathological studies showed that MVD, Ki-67, VEGF, and CD34 expression levels were significantly increased in the isolated tumor specimens of EAC. No impact of IM on the CA IX expression in vivo and in vitro was detected in this work. These facts led to the conclusion that co-administration of AZ can significantly extend the anti-angiogenic and anti-metastatic effects of IM therapy (Figure 6) [2923].
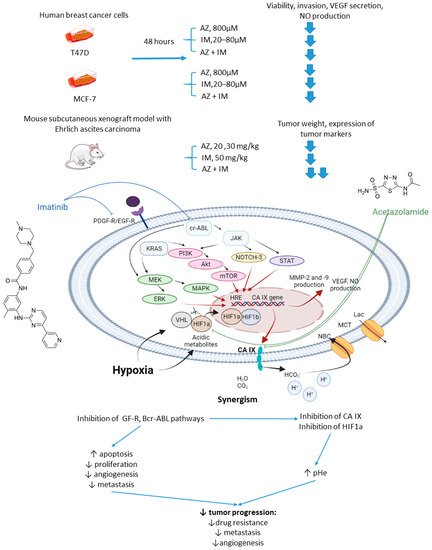
Figure 6. Graphical outline of the study on AZ+IM combination reported by Abd-El Fattah et al., and summary on the hypothesized molecular interactions resulting in the synergistic action of the drugs [31].
2.4. Acetazolamide and MS-275
Overexpression or aberrant recruitment of histone deacetylases (HDACs) to the promoter of tumor-suppressor genes is one of the most common epigenetic alterations in tumor onset and progression [3225]. Small molecule HDAC inhibitors, despite eliciting impressive results in preclinical settings, show limited efficacy in patients due to their toxic effects and emergence of drug resistance in cancer cells [3326]. Therefore, continuous efforts are being made to discover drug combinations that would allow for achieving the full therapeutic potential of HDAC inhibitors in patients [3427]. Of note, both CA IX and HDACs are overexpressed in neuroblastoma (Figure 7)[3528]. In the light of these facts, Mokhtarti and coworkers provided one comparing the efficacy of MS-275, a small-molecule HDAC inhibitor, AZ, and their combination within in vitro and in vivo models of neuroblastoma [3629]. Intriguingly, co-administration of AZ and MS-275 led to a stronger decrease in cell viability and migration capacity as compared to the monotherapies. In turn, exposure of neuroblastoma SH-SY5Y xenografts to the combined AZ+MS-275 treatment yielded a significant reduction in tumor growth and vascularization. It was reported a profound decrease in the expression of stemness markers in the tumors subjected to the AZ+MS-275 combination. Extensive apoptosis has been shown in the treated tumors, and the drug combination helped recover the T-cell balance. Finally, the combined treatment also led to a substantial downregulation of HIF1-α and CA IX, thereby confirming the contribution of AZ to the observed effects (Figure 7) [3629].
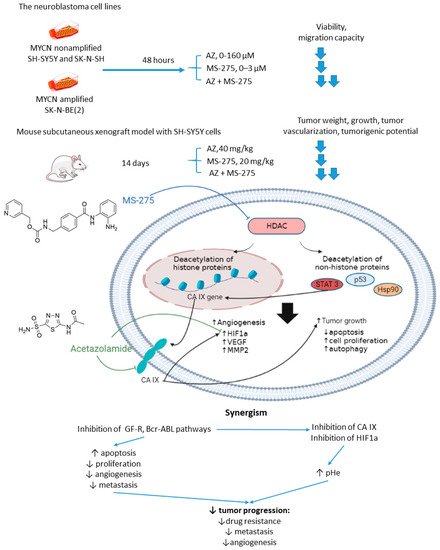
Figure 7. Graphical outline of the study on AZ+MS-275 combination reported by Mokharti et al., and summary on the hypothesized molecular interactions resulting in synergistic action of the drugs [37].
3. Acetazolamide in Multidrug Combinations
Acetazolamide and CHOP Combination
CHOP is a chemotherapy combination that is used to treat non-Hodgkin lymphoma [3830]. It includes cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate (Oncovin), and prednisone [3931]. Lymphomas are often marked with significant intratumor metabolic heterogenicity, which largely improves their therapy resistance [4032]. In particular, hypoxic and highly acidified compartments tend to show a poor response to CHOP medication [433].
4. Methazolamide in Combination with Conventional Cytostatic Agents
4.1. Methazolamide and Gemcitabine
Methazolamide and Gemcitabine
Joshi and colleagues employed another clinically used pan-isoform CAI methazolamide with a hope to intensify gemcitabine treatment of pancreatic cancers (Figure 8) [4134]. In the reported in vitro experiments, Joshi and coworkers tested methazolamide+gemcitabine drug combination against patient-derived pancreatic carcinoma cells PDX-1986, PDX-546, Capan-2, MIA PaCa-2, as well as immortalized PANC-1 cells cultured in 2D and 3D modes [4134]. Intriguingly, significant growth inhibition was produced by the drug combination compared to drug-alone controls. This encouraged to further test the combined medication regimen in PDX-546-bearing mice. Reassuringly, the combination group showed more significant tumor growth inhibition compared to the drug alone. Meanwhile, no detectable bodyweight loss suggesting good tolerance of the treatment. Histopathological studies revealed a pronounced reduction in expression of the stem cell markers and proliferation marker Ki-67 in the combination group, but not in drug-alone treated animals. In addition, both methazolamide and combination groups displayed anti-angiogenic morphological changes (would healing, tube formation), as well as a significant decrease in HIF1α and VEGF expression levels, highlighting the role of CAIs in the suppression of vascular growth in the tumor. Therefore, the potential of the methazolamide+gemcitabine combination for the treatment of pancreatic cancer has been preliminarily demonstrated (Figure 8) [4134].
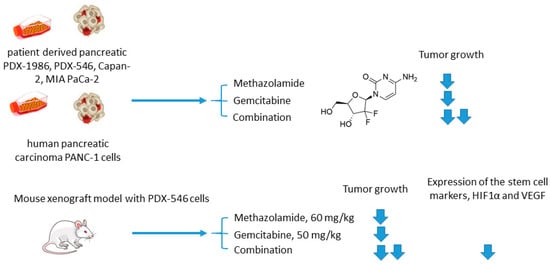
Figure 8. Graphical outline of the study on methazolamide+gemcitabine combination reported by Joshi et al. [42].
5. Isoform Selective Carbonic Anhydrase IX Inhibitors in Combination with Conventional Cytostatic Agents
5.1. SLC-0111 and Dacarbazine/Temozolomide/Doxorubicin/5-Fluorouracil
Andreucci and coworkers performed a detailed in vitro one on the potentiation of cytotoxic agents’ efficacy in the presence of SLC-0111, a CA IX isoform-selective inhibitor that recently entered Phase Ib/II clinical trials [4335]. To this end, human melanoma A375-M6, breast carcinoma MCF7, and colorectal carcinoma HCT 116 cell lines were employed.
It is demonstrated that SLC-0111 markedly augmented cell death percentage, late apoptosis, and necrosis in A375-M6 melanoma cells when co-administered with guanine methylating agents (dacarbazin or temozolomide). A similar effect was produced by the SLC-0111+DOX combination against MCF7 breast cancer cells. In addition, all combinations efficiently blocked cell colony formation. This was not true, however, for the combination of SLC-0111 and 5-fluorouracil, which did not affect the cell viability of HCT116 colorectal carcinoma cells. Thus, SLC-0111 displayed the potential to sensitize cancer cells to conventional cytostatic agents, which was especially true for weakly basic drugs, but not for the weak acid 5-fluorouracil (Figure 9) [4335].
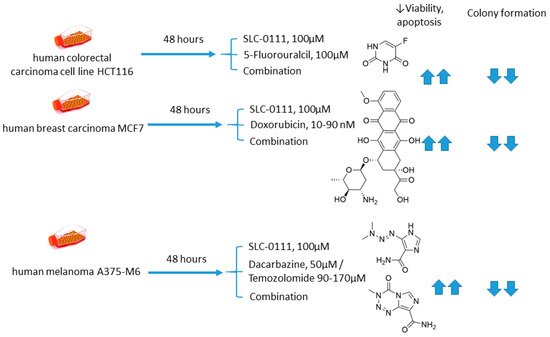
Figure 9. Graphical outline of the study on SLC-0111+dacarbazine/temozolomide/DOX/5-fluorouracil combination reported by Andreucci et al. [44].
5.2. SLC-0111 and Temozolomide
Boyd and colleagues investigated SLC-0111 for its ability to enhance the efficacy of temozolomide against glioblastoma in vitro and in vivo [4536]. It was revealed that monotherapy with SLC-0111 or temozolomide significantly decreased the growth of glioblastoma cells isolated from pediatric primary (D456) and a recurrent (1016 GBM) patient-derived xenograft in normoxia and hypoxia, whereas the combination caused a further drop in the cell growth but did not increase the toxicity of temozolomide against healthy astrocytes. It was shown that the SLC-0111+temozolomide combination prominently induced cell cycle arrest via DNA damage and lowered intracellular pH in cancer cells. Furthermore, the drug combination in question was highly efficient in vivo when administered to nude mice implanted with a 1016 GBM patient-derived xenograft. In fact, SLC-0111+temozolomide produced noticeable tumor regression in xenografts, and this effect was clearly greater than that of the drugs alone. Importantly, 130 days after xenograft implantation, the median survival for the temozolomide-alone treatment group was 76 days, while median survival could not be determined for the combination group. Analysis of tumor sections from the treated mice demonstrated that expression of the Brain Tumor Initiating Cell (BTIC) marker Sox2 was decreased with the co-administration of SLC-0111 with temozolomide, suggesting that SLC-0111 was capable of decreasing BTIC enrichment after temozolomide therapy. Therefore, the results obtained by Boyd highlight a significant benefit of the addition of SLC-0111 to the conventional temozolomide-based treatment for glioblastoma (Figure 10) [4536].
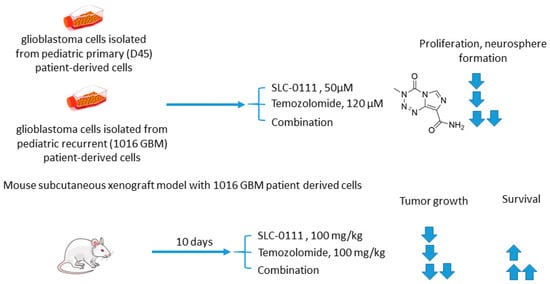
Figure 10. Graphical outline of the study on SLC-0111+temozolomide combination reported by Boyd et al. [46].
5.3. SLC-0111 and Gemcitabine
In 2019, McDonald and colleagues evaluated the potential of SLC-0111 to improve therapeutic outcomes in pancreatic ductal adenocarcinomas expressing an activated form of Ki-ras2 Kirsten rat sarcoma (KRAS) oncogene (Figure 11) [4737]. Approximately 93% of pancreatic adenocarcinomas harbor mutant KRAS oncogene, which drives tumor pathogenesis [4838]. Intriguingly, extensive in vitro and in vivo studies performed revealed that KRAS-driven pancreatic ductal adenocarcinomas display a dependency on glycolysis and the need to buffer intracellular pH through the bicarbonate producing activity of CA IX. Thus, CA IX was identified as a pharmacologically targetable vulnerability downstream of mutant KRAS, acting as a hypoxia/pH-specific effector induced by the oncogene. In light of these facts, it became of utmost interest to evaluate the potential of CA IX inhibitors to sensitize KRAS mutant cells to chemotherapeutic agents, specifically to gemcitabine, which is typically used against this type of cancer. McDonald and coworkers reported significant intracellular pH drop and decreased survival rates in pancreatic cancer cells exposed to SLC-0111+gemcitabine treatment. In CA IX-positive KRAS-mutant PaCa83–2 patient-derived xenografts, 16 weeks of administration of the drug combination resulted in a dramatic increase in survival, as 100% of mice given the combination were alive, with one animal remaining tumor-free after the treatment. Finally, a histopathological one of KrasG12D/Pdx1-Cre/p53/RosaYFP genetically engineered mouse model revealed that after 14 days of treatment, the combination group displayed significantly fewer B220+ B-cells compared to control and single agents, which can be considered beneficial in the context of recent reports on B-cells promoting pancreatic tumorigenesis [4939]. Meanwhile, no significant impact on the number of CD3+ T-cells was observed. Therefore, while suppressing tumor growth, glycolytic metabolic adaptation, and increasing survival rates in vivo, the drug combination did not have an adverse impact on the immune microenvironment (Figure 11) [4737].
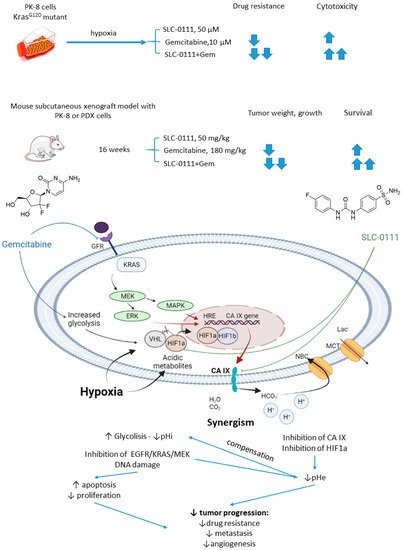
Figure 11. Graphical outline of the study on SLC-0111+gemcitabine combination reported by McDonald et al., and summary on the hypothesized molecular interactions resulting in synergistic action of the drugs [50].
5.4. S4 and Doxorubicin
Inspired by the acetazolamide-induced intensification of DOX treatment in CA IX-rich cell lines reported by Gieling et al. (vide supra), Kuijk and colleagues performed a follow-on one by using an isoform-selective CA IX inhibitor S4 [5140]. This ureido-substituted sulfamate SLC-0111 analog has been earlier described to exert significant antiproliferative efficacy in vitro in different breast cancer tumor models [5241][5342]. It should be noticed, however, that despite encouraging in vitro results, the in vivo efficacy of S4 remained ambiguous. Thus, S4 was ineffective in reducing primary tumor growth in vivo, although causing decreased spontaneous lung metastases formation in an orthotopic MDA-MB-231 breast cancer model [1543]. The S4 involving tests were carried out against MDA-MB-231 triple-negative breast adenocarcinoma, intact (CA IX high) as well as doxycycline-induced CA IX knockdown (CA IX low) HT-29 cells. S4-mediated CA IX inhibition increased DOX efficacy during hypoxia and normoxia exposure in MDA-MB-231 cells, compared to single drug exposure. These results were in line with the lack of S4 efficacy in the HT-29–CAIX low cells. However, contrary to what was expected, DOX sensitivity decreased when HT-29–CA IX low cells were exposed to S4 during hypoxic conditions. Higher serum concentrations also abrogated the effect of S4 on DOX efficacy, which may occur due to the high binding affinity of S4 to bovine serum albumin. MDA-MB-231 tumor-bearing mice were consequently treated with the DOX+S4 combination to evaluate its in vivo efficacy. Disappointingly, S4 co-administration abrogated the effect of DOX in this animal model, and the reasons remained unclear [5140].
5.5. S4 and Cisplatin
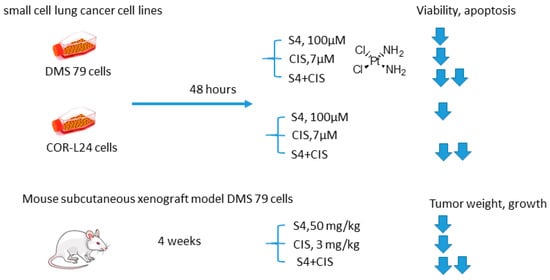
Soon thereafter, it is on S4+CIS combination against small cell lung cancer emerged (Figure 12) [1644]. Bryant and coworkers reported that S4+CIS reduced cell viability in two human small cell lung cancer DMS-79 COR-L24 cell lines. Moreover, in DMS-79 xenograft tumors in nude mice, the S4+CIS combination significantly delayed the tumor growth. Of note is a profound decrease in the necrotic area (almost 50% of tumor area was comprised of necrosis) in the combination therapy group. Dosing with CIS after completion of the 3-week schedule of S4 resulted in a response equal to that of chemo-naive tumors. Therefore, no resistance to therapy was acquired during this treatment. In turn, COR-L24 xenograft tumors showed exquisite sensitivity to S4, keeping tumor sizes at ca. 250 mm3 for the 4-week schedule of S4. COR-L24 xenograft tumors were also more CIS responsive than DMS 79, but the treatment was poorly tolerated. In this model, the combination of therapies produced a slightly better treatment response than single agents with COR-L24 tumor regression in three out of four mice showing a strong response. The histological analysis highlighted a significant reduction in CA IX expression and reduced hypoxia regions in COR-L24 tumor xenografts in response to S4 treatment (Figure 12) [1644].
5.6. n-Octyl Disulfamate/SCL-0111 and 3-O-acetylbetulin
Petrenko and colleagues, in 2021, it was presented that on a combination of CA IX inhibitor n-octyl disulfamate (OCT) with a pentacyclic triterpene 3-O-acetylbetulin (3-AC), a betulinic acid prodrug that shows selective cytostatic activity on human cancer cell lines in vitro and in vivo (Figure 13) [5645]. When co-administered, OCT and 3-AC produced remarkable antiproliferative activity and inhibited cell migration in MDA-MB-231 and MCF-7 breast cancer cell lines. Both effects were significantly higher for the combined treatment than the single-drug regimens. Subsequently, SLC-0111 was also tested with 3-AC against breast cancer cells. This drug combination produced a considerable drop in clonogenic survival rates of MDA-MB-231 and MCF-7 cell cultures. Additionally, in MDA-MB-231, but not in MCF-7 cell cultures, increased cells sensitivity towards radiotherapy was observed under exposure to SLC-0111+3-AC treatment (Figure 13) [5645].
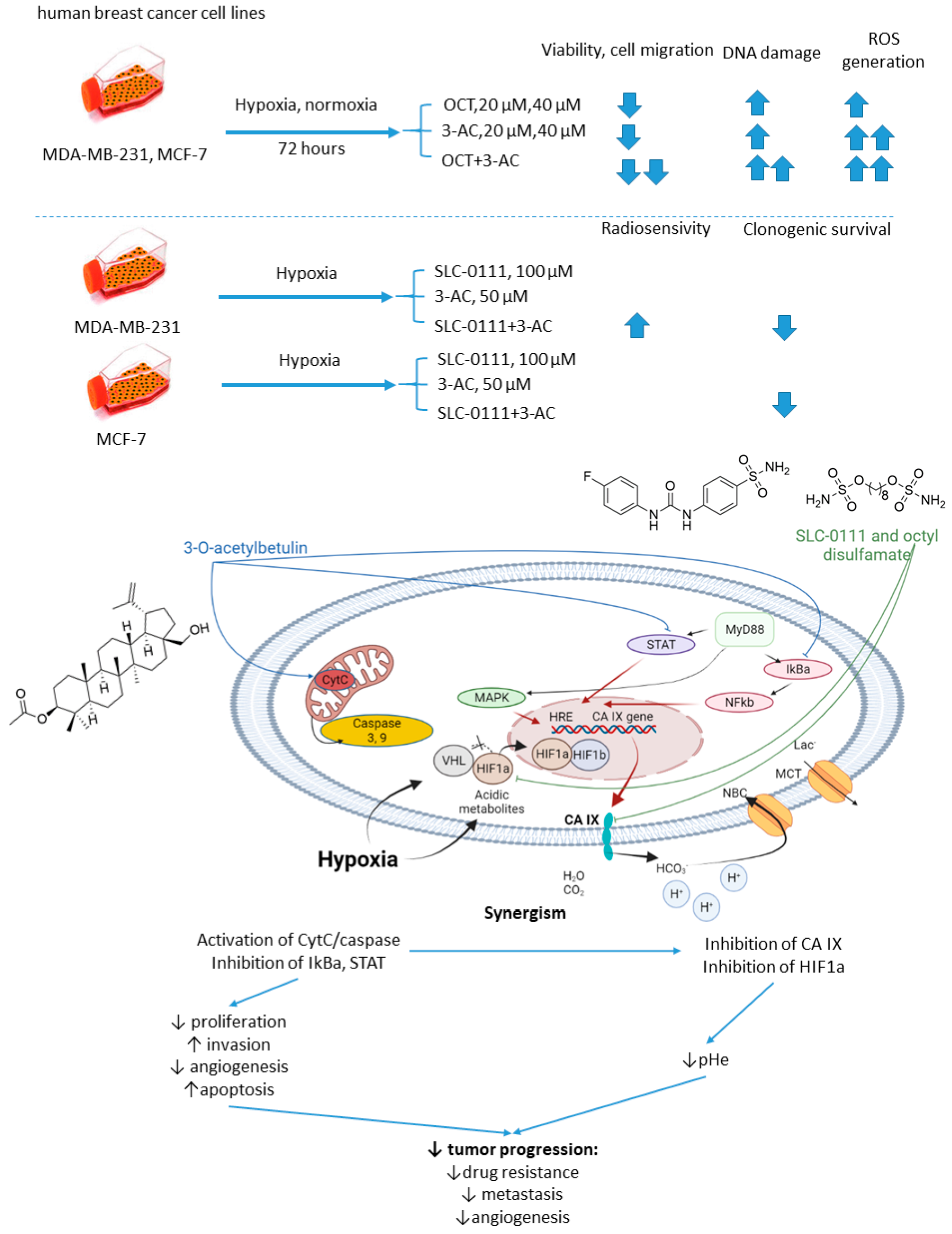
Figure 13. Graphical outline of the study on SLC-0111/OCT+3-AC combination reported by Petrenko et al., and summary on the hypothesized molecular interactions resulting in synergistic action of the drugs [57].
Abbreviations
| AZ | acetazolamide |
| BTIC | brain tumor initiating cell |
| CA | carboanhydrase |
| CA IX | carbonic anhydrase IX |
| CAIs | CA inhibitors |
| CHOP | cyclophosphamide, doxorubicin hydrochloride hydroxydaunorubicin, vincristine sulfate Oncovin, and prednisone |
| CIS | cisplatin |
| DOX | doxorubicin |
| EAC | Ehrlich ascites carcinoma |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-regulated kinase |
| EV1 | empty vector |
| GFR | growth factor receptor |
| HDAC | histone deacetylases |
| HUVEC | human umbilical vein endothelial cells |
| IM | imatinib |
| JAK | Janus kinase |
| KEAP1 | Kelch Like ECH Associated Protein 1 |
| KRAS | Kirsten rat sarcoma |
| MAPK | mitogen activated protein kinase |
| MEK | MAPK/ERK kinase |
| MMP | matrix metalloproteinases |
| mTOR | mechanistic target of rapamycin |
| mTORC | mTOR complexe |
| OCT | n-octyl disulfamate |
| PDGFR | platelet-derived growth factor receptor |
| PDK1 | pyruvate dehydrogenase kinase 1 |
| PGK1 | phosphoglycerate kinase 1 |
| pHe | extracellular pH |
| pHi | intracellular pH |
| PI3K | phosphatidylinositol-3 kinase |
| PPIs | proton pump inhibitors |
| SFN | sulforaphane |
| TIMP-1 | tissue inhibitor of metalloprotease-1 |
| VEGF | vascular endothelial growth factor |