Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Chang Yong Oh and Version 2 by Bruce Ren.
DNA nanoengineering, in particular, DNA origami has potential applications in a variety of areas including, for example, nanoelectronics, biomedical diagnostics, and therapeutics. To fully realize the potential of DNA self-assembly in these and other areas, methods must be available for economical, scalable, and reliable production of single-stranded DNA (ssDNA) scaffolds from virtually any source. Described here are four such methods.
- DNA nanoengineering
- DNA origami
- scaffold
- rolling circle amplification
- lambda exonuclease
- chemical denaturation
- asymmetric polymerase chain reaction
1. Introduction
Nanoengineering by virtue of DNA-based self-assembly [1][2][3][4][5] is emerging as a platform methodology for addressing a variety of interesting issues ranging from nanoelectronics to biomedical diagnostics and therapeutics [5][6][7][8][9][10]. In many (but not all) of these methods, a single-stranded nucleic acid “scaffold” is required, and this role is often played by the natural single-stranded DNA (ssDNA) scaffold of the bacteriophage M13, predominately because of its accessibility [3][11]. Although the use of the M13 scaffold has led to a vast number of advances, it significantly constrains the realization of the full potential of the DNA self-assembly platform. Thus, to fully leverage the inherent utility of DNA nanoengineering it must be possible to create ssDNA scaffolds easily and reliably from essentially any source [11][12][13]. The researchers describe the virtues and liabilities of four relatively economical and simple approaches for producing long ssDNA molecules. These methods are 1, Rolling Circle Amplification (RCA), 2, strand-specific exonuclease digestion, 3, chemical denaturation, and, 4, asymmetric polymerase chain reaction (aPCR).
1.1. Rolling Circle Amplification (RCA)
Rolling Circle Amplification (RCA) is a very efficient method for generating large quantities of ssDNA in a simple isothermal reaction [11][14][15][16]. Standard RCA employs phi29 DNA polymerase because of its high processivity and strong strand displacement ability [11][14][17]. As such, this method is capable of producing long concatemers of the desired DNA sequence [11][16][17]. Further, this approach can be scaled to industrial levels [16]. However, standard RCA carried out using phi29 polymerase has the intrinsic problem of producing terminal double-stranded DNA (dsDNA) byproducts by consuming ssDNA products in what has been described as a “strand jumping” event. To mitigate this situation, single-strand binding protein (SSB) can be included in the RCA reaction, but at additional cost and creating a need for additional post-synthesis purification steps [17]. Also, the reaction requires a circular template, nicked on the desired strand, which can be problematic in some instances [18] since the substrate for the reaction must contain one or more sites amenable to nicking by a relatively small pool of sequence-specific nicking endonucleases (“nickases”) [16][17]. Finally, cleavage of concatemeric RCA products into monomeric ssDNA is critical for the utilization of products for downstream applications [19]. Thus, for the efficient production of large quantities of ssDNA at a low cost, RCA is only applicable in a subset of cases in which these favorable conditions exist.
1.2. Strand-Specific Lambda Exonuclease Digestion
Lambda exonuclease preferentially digests dsDNA from the 5′-phosphorylated end. This feature can be leveraged to create ssDNA by asymmetrically phosphorylating the 5′ end of a dsDNA template, generally prepared by PCR [11][20][21][22]. It is noteworthy that lambda exonuclease is also capable of digesting the non-phosphorylated 5′-OH strand, but at a lower, albeit, not insignificant rate [21][22]. Thus, the enzyme concentration and incubation time must be carefully optimized to produce predominantly full-length desired strand [21][22]. A useful modification of this method to rectify this inherent problem is to asymmetrically modify the 5′ termini to protect the desired strand in concert with the 5′ phosphorylation of the undesired strand. Protective modifications include, for example, the addition of 5′ inverted dT, the inclusion of terminal phosphorothioate bonds, and 5′ biotinylation [22][23]. Terminal base modifications have to be chosen carefully based on downstream applications since they have the potential to impact the behavior of the resultant modified ssDNA. Phosphorothioate modification has been reported to be relatively innocuous in this respect [23][24]. Coupled with chemical protection of the 5′ terminus of the desired strand, strand-specific lambda exonuclease digestion offers the benefits of rapid production and optimization [20][21][22]. Despite the simplicity and rapid production, the strand-specific lambda degradation method may be sub-optimal for large-scale production of ssDNA. Incomplete degradation of dsDNA contributes to low production yield and the purification steps required for removal of enzyme often result in further loss of ssDNA [11][21]. In some cases, these drawbacks can be alleviated by coupling this approach with well-optimized conventional PCR and ensuring complete 5′ phosphorylation of the target strand [22].
1.3. Chemical Denaturation
Chemicals such as urea, formamide, and sodium hydroxide (NaOH) can be used to denature the DNA double helix via hydrogen bond disruption [21][25][26][27][28][29]. Identification and isolation of the desired strand post-denaturation can be accomplished by, for example, asymmetric biotin modification using a variety of streptavidin (or avidin) selection systems [21][25][26] or agarose gel electrophoresis (AGE) [24][26][27]. NaOH and urea are the most commonly used chemical denaturants based on their high efficiency, and economy [21][27][28][30][31]. However, these methods can be somewhat dangerous, and extensive purification may be required to remove residual denaturants [21][26][28]. Further, when coupled with biotin-streptavidin selection systems, the NaOH concentration has to be carefully chosen to obtain the maximum desired product yield [30]. Thus, the researchers have developed a simpler method wherein the dsDNA is briefly heated in a 50% formamide solution [25][29] and rapidly loaded on a native agarose gel, thereby minimizing the re-hybridization of the two strands. It is advantageous to nick the undesired strand with a commercially available nicking endonuclease to reduce the probability that the two ssDNA strands will co-migrate and thereby preclude facile isolation of the desired strand [27]. Following denaturation and electrophoretic strand separation, the desired strand must be removed from the gel which, with existing commercial methods, can reduce yield.
1.4. Asymmetric PCR (aPCR)
Asymmetric PCR (aPCR) is one of the simplest methods for generating ssDNA from a dsDNA template, the latter usually generated by standard PCR [11]. aPCR produces an asymmetric distribution of the two strands by using unequal concentrations of forward and reverse primers, one of which is present, by choice, in a limiting concentration. Initially, dsDNA is produced but once the limiting primer is consumed there is a shift to linear (non-logarithmic) production of the desired ssDNA by the primer that is present in excess [11][32][33]. Despite its apparent simplicity, aPCR can be fraught with sequence-based limitations (e.g., primer site duplicity, excessive length, byproduct formation by ssDNA product dimerization). Thus, careful optimization is required for each template and in some cases, a satisfactory parameter set cannot be readily established [33]. Methods have been developed to help alleviate this problem. For example, Veneziano et al. compared a wide range of commercially available thermally stable DNA polymerases and identified several that demonstrate superior ability to produce large-scale ssDNA by aPCR [32].
2.Comparison of Methods for the Production of Kilobase-Length Single-Stranded DNA
1. Introduction
Nanoengineering by virtue of DNA-based self-assembly [1,2,3,4,5] is emerging as a platform methodology for addressing a variety of interesting issues ranging from nanoelectronics to biomedical diagnostics and therapeutics [5,6,7,8,9,10]. In many (but not all) of these methods, a single-stranded nucleic acid “scaffold” is required, and this role is often played by the natural single-stranded DNA (ssDNA) scaffold of the bacteriophage M13, predominately because of its accessibility [3,11]. Although the use of the M13 scaffold has led to a vast number of advances, it significantly constrains the realization of the full potential of the DNA self-assembly platform. Thus, to fully leverage the inherent utility of DNA nanoengineering it must be possible to create ssDNA scaffolds easily and reliably from essentially any source [11,12,13]. In this report, we describe the virtues and liabilities of four relatively economical and simple approaches for producing long ssDNA molecules. These methods are 1, Rolling Circle Amplification (RCA), 2, strand-specific exonuclease digestion, 3, chemical denaturation, and, 4, asymmetric polymerase chain reaction (aPCR).
1.1. Rolling Circle Amplification (RCA)
Rolling Circle Amplification (RCA) is a very efficient method for generating large quantities of ssDNA in a simple isothermal reaction [11,14,15,16]. Standard RCA employs phi29 DNA polymerase because of its high processivity and strong strand displacement ability [11,14,17]. As such, this method is capable of producing long concatemers of the desired DNA sequence [11,16,17]. Further, this approach can be scaled to industrial levels [16]. However, standard RCA carried out using phi29 polymerase has the intrinsic problem of producing terminal double-stranded DNA (dsDNA) byproducts by consuming ssDNA products in what has been described as a “strand jumping” event. To mitigate this situation, single-strand binding protein (SSB) can be included in the RCA reaction, but at additional cost and creating a need for additional post-synthesis purification steps [17]. Also, the reaction requires a circular template, nicked on the desired strand, which can be problematic in some instances [18] since the substrate for the reaction must contain one or more sites amenable to nicking by a relatively small pool of sequence-specific nicking endonucleases (“nickases”) [16,17]. Finally, cleavage of concatemeric RCA products into monomeric ssDNA is critical for the utilization of products for downstream applications [19]. Thus, for the efficient production of large quantities of ssDNA at a low cost, RCA is only applicable in a subset of cases in which these favorable conditions exist.
1.2. Strand-Specific Lambda Exonuclease Digestion
Lambda exonuclease preferentially digests dsDNA from the 5′-phosphorylated end. This feature can be leveraged to create ssDNA by asymmetrically phosphorylating the 5′ end of a dsDNA template, generally prepared by PCR [11,20,21,22]. It is noteworthy that lambda exonuclease is also capable of digesting the non-phosphorylated 5′-OH strand, but at a lower, albeit, not insignificant rate [21,22]. Thus, the enzyme concentration and incubation time must be carefully optimized to produce predominantly full-length desired strand [21,22]. A useful modification of this method to rectify this inherent problem is to asymmetrically modify the 5′ termini to protect the desired strand in concert with the 5′ phosphorylation of the undesired strand. Protective modifications include, for example, the addition of 5′ inverted dT, the inclusion of terminal phosphorothioate bonds, and 5′ biotinylation [22,23]. Terminal base modifications have to be chosen carefully based on downstream applications since they have the potential to impact the behavior of the resultant modified ssDNA. Phosphorothioate modification has been reported to be relatively innocuous in this respect [23,24]. Coupled with chemical protection of the 5′ terminus of the desired strand, strand-specific lambda exonuclease digestion offers the benefits of rapid production and optimization [20,21,22]. Despite the simplicity and rapid production, the strand-specific lambda degradation method may be sub-optimal for large-scale production of ssDNA. Incomplete degradation of dsDNA contributes to low production yield and the purification steps required for removal of enzyme often result in further loss of ssDNA [11,21]. In some cases, these drawbacks can be alleviated by coupling this approach with well-optimized conventional PCR and ensuring complete 5′ phosphorylation of the target strand [22].
1.3. Chemical Denaturation
Chemicals such as urea, formamide, and sodium hydroxide (NaOH) can be used to denature the DNA double helix via hydrogen bond disruption [21,25,26,27,28,29]. Identification and isolation of the desired strand post-denaturation can be accomplished by, for example, asymmetric biotin modification using a variety of streptavidin (or avidin) selection systems [21,25,26] or agarose gel electrophoresis (AGE) [24,26,27]. NaOH and urea are the most commonly used chemical denaturants based on their high efficiency, and economy [21,27,28,30,31]. However, these methods can be somewhat dangerous, and extensive purification may be required to remove residual denaturants [21,26,28]. Further, when coupled with biotin-streptavidin selection systems, the NaOH concentration has to be carefully chosen to obtain the maximum desired product yield [30]. Thus, we have developed a simpler method wherein the dsDNA is briefly heated in a 50% formamide solution [25,29] and rapidly loaded on a native agarose gel, thereby minimizing the re-hybridization of the two strands. It is advantageous to nick the undesired strand with a commercially available nicking endonuclease to reduce the probability that the two ssDNA strands will co-migrate and thereby preclude facile isolation of the desired strand [27]. Following denaturation and electrophoretic strand separation, the desired strand must be removed from the gel which, with existing commercial methods, can reduce yield.
1.4. Asymmetric PCR (aPCR)
Asymmetric PCR (aPCR) is one of the simplest methods for generating ssDNA from a dsDNA template, the latter usually generated by standard PCR [11]. aPCR produces an asymmetric distribution of the two strands by using unequal concentrations of forward and reverse primers, one of which is present, by choice, in a limiting concentration. Initially, dsDNA is produced but once the limiting primer is consumed there is a shift to linear (non-logarithmic) production of the desired ssDNA by the primer that is present in excess [11,32,33]. Despite its apparent simplicity, aPCR can be fraught with sequence-based limitations (e.g., primer site duplicity, excessive length, byproduct formation by ssDNA product dimerization). Thus, careful optimization is required for each template and in some cases, a satisfactory parameter set cannot be readily established [33]. Methods have been developed to help alleviate this problem. For example, Veneziano et al. compared a wide range of commercially available thermally stable DNA polymerases and identified several that demonstrate superior ability to produce large-scale ssDNA by aPCR [32].
2.Comparison of Methods for the Production of Kilobase-Length Single-Stranded DNA
Figure 1 is a schematic diagram of each of the methods described above. Standard PCR was used to prepare templates for lambda exonuclease, formamide separation, and aPCR. Verification and purification in all cases were carried out by AGE and the use of a commercially available gel recovery kit as described in Materials and Methods. Verification of ssDNA products from each method was carried out by restriction enzyme digestion and SSB treatment.
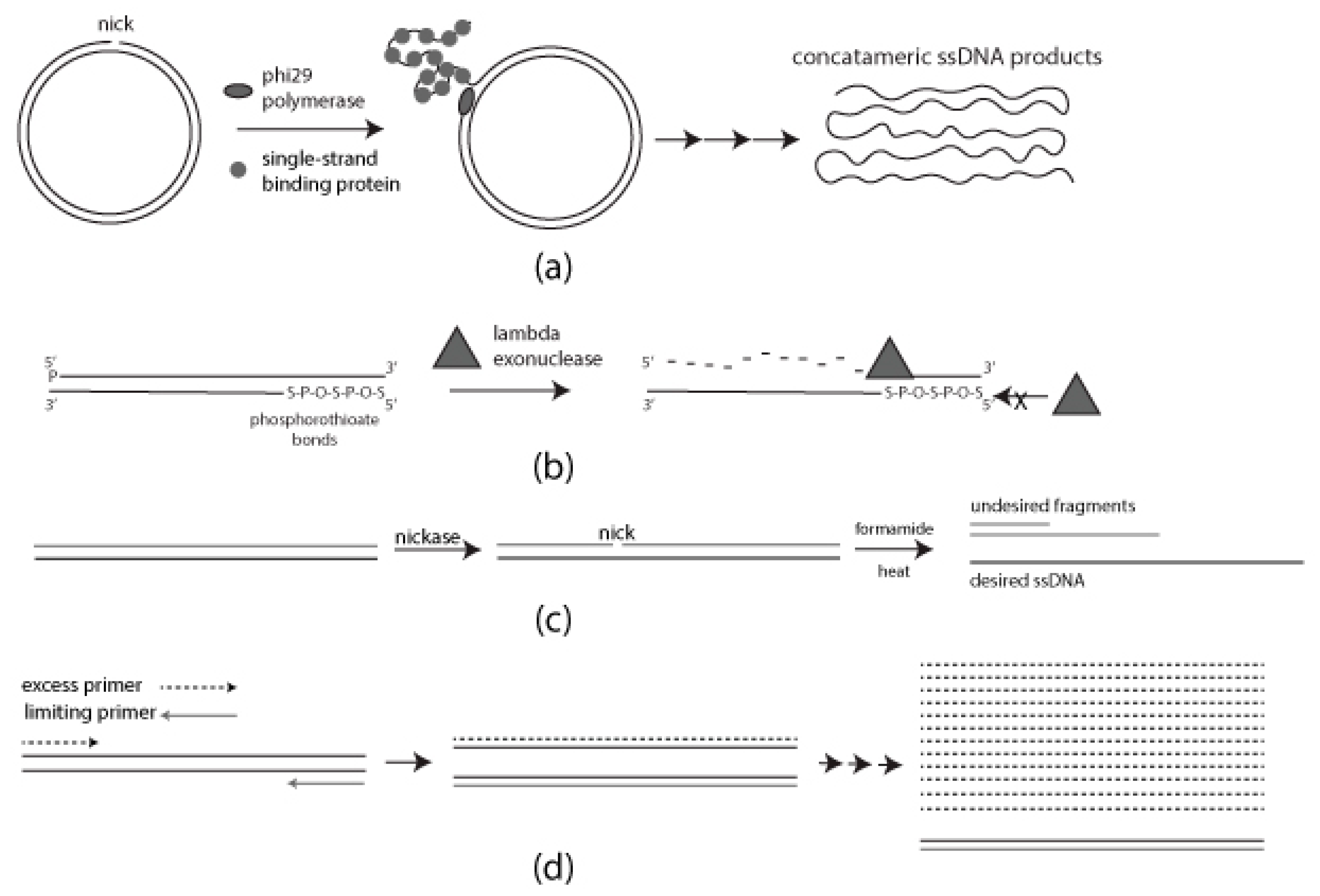
is a schematic diagram of each of the methods described above. Standard PCR was used to prepare templates for lambda exonuclease, formamide separation, and aPCR. Verification and purification in all cases were carried out by AGE and the use of a commercially available gel recovery kit as described in Materials and Methods. Verification of ssDNA products from each method was carried out by restriction enzyme digestion and SSB treatment.
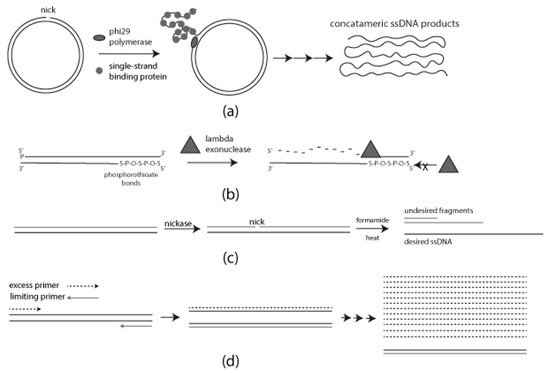
Figure 1.
Schematic diagram of each method for generating single-stranded DNA (ssDNA): (
a
) Rolling Circle Amplification (RCA) requires single-strand binding protein (SSB) for a progressive generation of single-stranded DNA (ssDNA); (
b
) Selective digestion of 5′ phosphorylated strand mediated by lambda exonuclease. Phosphorothioate bonds on the desired strand for protection; (
c
) Formamide-directed strand separation and strand selection through nicking endonuclease treatment and Agarose gel electrophoresis (AGE); (
d) Use of unequal concentrations of forward and reverse primers to generate ssDNA.
2.1. Rolling Circle Amplification (RCA)
RCA in the presence and absence of SSB resulted in high molecular weight bands above 10 kb and material retained in the gel wells. Restriction digestion was used to evaluate RCA products. In the case of RCA without SSB, the high molecular weight band above 10 kb was fully digested whereas the high molecular weight material in the gel wells remained undigested, suggesting that a significant portion of RCA product in the absence of SSB was dsDNA with some high molecular weight ssDNA also produced. In the presence of SSB, both high molecular weight bands above 10 kb and in the gel wells were resistant to digestion, indicating that they were predominantly ssDNA () Use of unequal concentrations of forward and reverse primers to generate ssDNA.
2.1. Rolling Circle Amplification (RCA)
RCA in the presence and absence of SSB resulted in high molecular weight bands above 10 kb and material retained in the gel wells. Restriction digestion was used to evaluate RCA products. In the case of RCA without SSB, the high molecular weight band above 10 kb was fully digested whereas the high molecular weight material in the gel wells remained undigested, suggesting that a significant portion of RCA product in the absence of SSB was dsDNA with some high molecular weight ssDNA also produced. In the presence of SSB, both high molecular weight bands above 10 kb and in the gel wells were resistant to digestion, indicating that they were predominantly ssDNA (
Figure 2a).

a).

Figure 2.
AGE results from each method described here for generating ssDNA. (
a
) RCA results in presence or absence of SSB. In the absence of SSB the primary product is double-stranded DNA (dsDNA). (
b
) Comparison of selective digestion of the 5′ phosphorylated strand of a linear DNA duplex by lambda exonuclease for 30, 60, 120, and 240 min. (
c
) Formamide-mediated strand separation following nicking endonuclease treatment of a linear DNA duplex. Nicking of the undesired strand greatly improves resolution of the desired strand. (
d) Generation of ssDNA by asymmetric PCR (aPCR). SS: ssDNA; DS: dsDNA.
2.2. Strand-Specific Lambda Exonuclease Degradation
Templates for lambda exonuclease-directed degradation were 5′-phosphorylated on the undesired (GFP sense) strand. Conversely, five phosphorothioate linkage modifications on 5′ terminus on the desired (GFP antisense) strand. This arrangement favors selective degradation of the undesired strand. To determine the optimum digestion time for lambda-directed degradation, four reactions were performed for 30, 60, 120, and 240 min and the results were visualized by AGE. In all four reactions, two bands were observed including one ~1.5 kb and another ~900 bp. Judging from the differences in mobility and staining efficiency, and corroborated by atomic force microscopy (AFM), the band at approximately 900 bp was single-stranded Green Fluorescent Protein DNA (ssGFP). Extending the reaction time led to further digestion of the dsGFP template but also some digestion of the desired ssDNA strand () Generation of ssDNA by asymmetric PCR (aPCR). SS: ssDNA; DS: dsDNA.
2.2. Strand-Specific Lambda Exonuclease Degradation
Templates for lambda exonuclease-directed degradation were 5′-phosphorylated on the undesired (GFP sense) strand. Conversely, five phosphorothioate linkage modifications on 5′ terminus on the desired (GFP antisense) strand. This arrangement favors selective degradation of the undesired strand. To determine the optimum digestion time for lambda-directed degradation, four reactions were performed for 30, 60, 120, and 240 min and the results were visualized by AGE. In all four reactions, two bands were observed including one ~1.5 kb and another ~900 bp. Judging from the differences in mobility and staining efficiency, and corroborated by atomic force microscopy (AFM), the band at approximately 900 bp was single-stranded Green Fluorescent Protein DNA (ssGFP). Extending the reaction time led to further digestion of the dsGFP template but also some digestion of the desired ssDNA strand (
Figure 2b). Thus, prolonging the reaction time to fully remove the remaining dsDNA fraction was not pursued since it reduced the overall yield of the desired product and potentially compromised its full-length integrity. This method is relatively simple and provides good yield but has the caveats of incomplete processing (in the interest of preserving full-length ssDNA) and subsequent reduction in final product yield due to losses during post agarose gel purification using commercial selective-filter-based systems.
2.3. Chemical Denaturation
dsGFP template prepared by conventional PCR was used in chemical denaturation (formamide) studies. The dsGFP was nicked on the undesired strand using the nickase Nt.BsmAI. Nicked products were composed of three different size fragments: a 1676 bp fully intact desired strand, and 1249 bp and 427 bp undesired strand fragments. Nicked dsGFP was purified by AGE followed by gel extraction using a commercially available kit as described in Materials and Methods. Purified nicked products were treated with 2× 95% formamide dye (Thermofisher) and heated to 80 °C to fully denature the duplex. The single strands were separated by AGE using a native agarose gel with minimal cooling permitted during loading the gel. Each separated fragment was identified based on its mobility on the gel. The AGE results showed that, as expected, a fraction of presumably re-hybridized dsDNA was present. Nonetheless, a substantial fraction remained single-stranded, and the desired strand was clearly separated from the smaller undesired strand fragments. It is noteworthy that isolation of the desired ssDNA strand was challenging in this instance due to the relatively small separation distance between the desired strand and the larger of the two undesired strand fragments (b). Thus, prolonging the reaction time to fully remove the remaining dsDNA fraction was not pursued since it reduced the overall yield of the desired product and potentially compromised its full-length integrity. This method is relatively simple and provides good yield but has the caveats of incomplete processing (in the interest of preserving full-length ssDNA) and subsequent reduction in final product yield due to losses during post agarose gel purification using commercial selective-filter-based systems.
2.3. Chemical Denaturation
dsGFP template prepared by conventional PCR was used in chemical denaturation (formamide) studies. The dsGFP was nicked on the undesired strand using the nickase Nt.BsmAI. Nicked products were composed of three different size fragments: a 1676 bp fully intact desired strand, and 1249 bp and 427 bp undesired strand fragments. Nicked dsGFP was purified by AGE followed by gel extraction using a commercially available kit as described in Materials and Methods. Purified nicked products were treated with 2× 95% formamide dye (Thermofisher) and heated to 80 °C to fully denature the duplex. The single strands were separated by AGE using a native agarose gel with minimal cooling permitted during loading the gel. Each separated fragment was identified based on its mobility on the gel. The AGE results showed that, as expected, a fraction of presumably re-hybridized dsDNA was present. Nonetheless, a substantial fraction remained single-stranded, and the desired strand was clearly separated from the smaller undesired strand fragments. It is noteworthy that isolation of the desired ssDNA strand was challenging in this instance due to the relatively small separation distance between the desired strand and the larger of the two undesired strand fragments (
Figure 2c). This can be rectified by a longer gel run and/or the use of a different nickase in many cases. The method is simple and rapid although for the sample chosen for this study (the GFP gene) the results were compromised by the presence of residual dsDNA and close migration rates of the desired and the larger of the two undesired ssDNA fragments.
2.4. Asymmetric PCR (aPCR)
aPCR was carried out using the forward and reverse primers used in standard PCR but at asymmetric ratios. Electrophoretic analysis of the aPCR reaction revealed that large quantities of both dsDNA and ssDNA were created by this method (c). This can be rectified by a longer gel run and/or the use of a different nickase in many cases. The method is simple and rapid although for the sample chosen for this study (the GFP gene) the results were compromised by the presence of residual dsDNA and close migration rates of the desired and the larger of the two undesired ssDNA fragments.
2.4. Asymmetric PCR (aPCR)
aPCR was carried out using the forward and reverse primers used in standard PCR but at asymmetric ratios. Electrophoretic analysis of the aPCR reaction revealed that large quantities of both dsDNA and ssDNA were created by this method (
Figure 2d). Unfortunately, aPCR conditions for all potential substrates must be carefully optimized and, in some cases, ideal conditions may not be obtainable. Further, ssDNA produced by this method must be efficiently separated and isolated from agarose gels and this can also be problematic with common commercial DNA isolation systems (i.e., yields can be unacceptably low).
d). Unfortunately, aPCR conditions for all potential substrates must be carefully optimized and, in some cases, ideal conditions may not be obtainable. Further, ssDNA produced by this method must be efficiently separated and isolated from agarose gels and this can also be problematic with common commercial DNA isolation systems (i.e., yields can be unacceptably low).