The incidence of non-viral causes of hepatocellular carcinoma, such as non-alcoholic steatohepatitis (NASH), is rising. The introduction of immune checkpoint inhibitors (ICI) has led to a paradigm shift in the systemic treatment of HCC. However, not all patients can benefit from ICI. Studies have suggested that the response to ICI may allude to the underlying aetiology of HCC, such as NASH.
- hepatocellular carcinoma
- NASH-HCC
- immune checkpoint inhibitors
1. Introduction
2. Immune Surveillance, Immune Microenvironment, and the Immune Checkpoints
2.1. Immunoediting
2.2. The Immune Microenvironment in the Liver
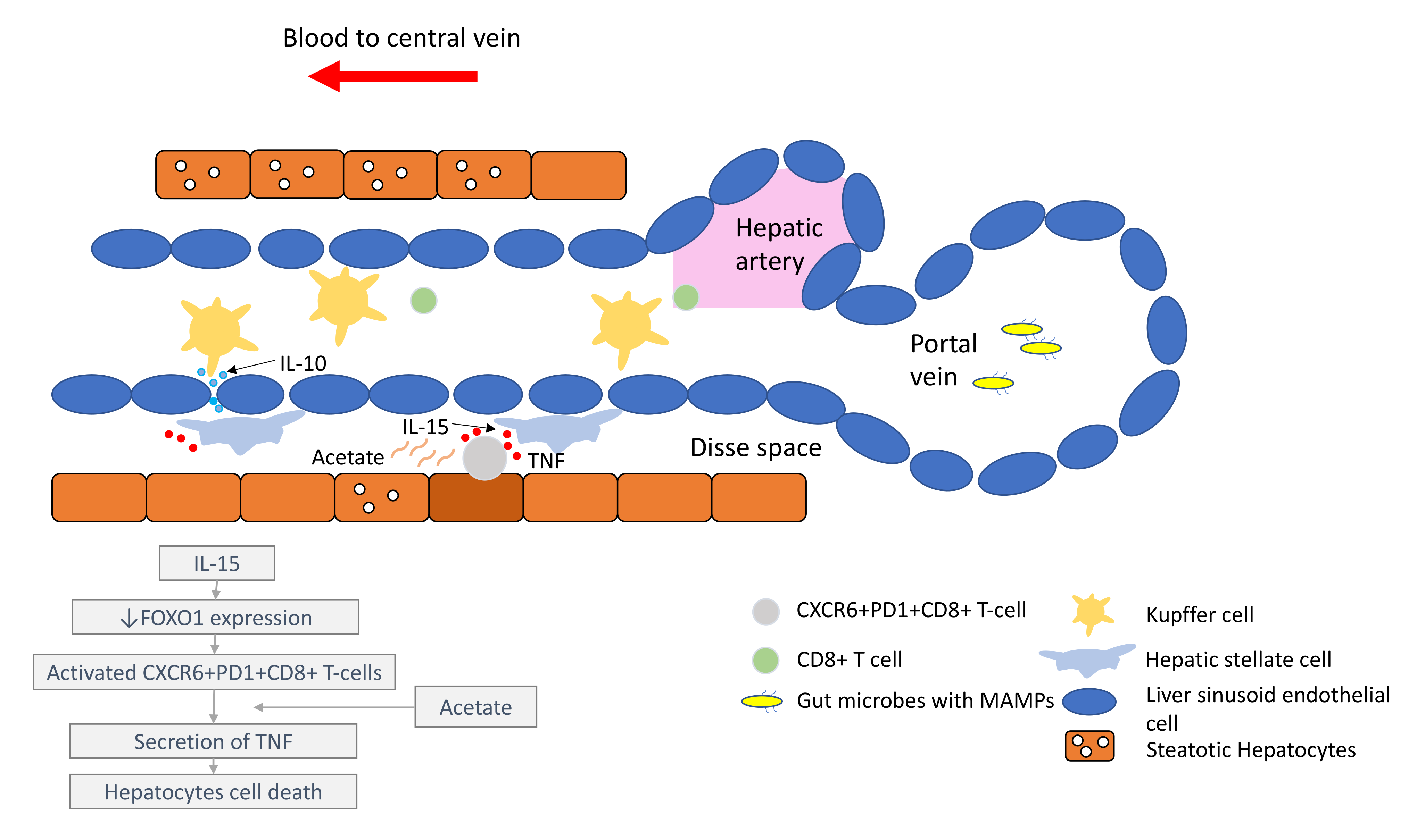
It is important to understand that the pathogenesis of HCC, regardless of the underlying aetiology, stems from chronic inflammation. It is essential to recognise that the immune microenvironment in the liver is distinctive to other organs, in order to prioritise biomarkers for drug development and treatment strategies. The liver has a unique blood supply, with the hepatic arterial blood joining the portal venous blood and draining into the hepatic veins, within a structure called the liver sinusoid. A large spectrum of microbes, microbe-associated molecular patterns (MAMPs) and damage-associated molecular patterns (DAMPs) continuously shower the liver sinusoids from the gut through the portal blood [7]. While these foreign molecules are constantly recognised and removed by tissue residential immune cells, such as the Kupffer cells (KCs), immunotolerance is maintained through the intricate interactions between the basal pro-inflammatory molecules (e.g., IL-2, IL-7, IL-12, IL-15 and IFN-γ) produced by the hepatic stellate cells (HSCs), NK cells, NKT-cells and γδ T-cells, and counter-balanced by anti-inflammatory cytokines (e.g., IL-10, IL-13, transforming growth factor beta (TGF-β)) produced by the myeloid-derived suppressor cells (MDSCs), regulatory T-cells (Tregs), liver sinusoidal endothelial cells (LSEC) and KCs [8][9].
Under conditions of chronic inflammation, this balance is lost and tips towards the pro-inflammatory state, resulting in increased hepatocytes turnover, compensatory proliferation, acquisition of mutations and malignant changes [9]. Eventually, HCC develops under the background of exhausted and dysfunctional effector cytotoxic T-cells. This is accompanied by the immunosuppressive tumour microenvironment, which is bathed with tumour-associated macrophages (TAMs), Tregs and MDSCs.
Comprehensive multiomics and single-cell analyses of human HCC tissues provided support of the presence of immunosuppressive cell populations in the tumour microenvironment, which fostered the growth of HCC [10][11]. Amongst them, TAMs are the most well studied. TAMs consist of KCs and blood/bone borne monocyte-derived macrophages. TAMs promote HCC progression in several ways, including the secretion of IL-10 and other immunosuppressive cytokines, promotion of angiogenesis, recruitment of Tregs and IL-17-expressing CD4+ T helper 17 cells, expression of inhibitory immune-checkpoint ligand PD-L1 and induction of proliferation signalling pathways [9][12][13]. Together with a high number of MDSCs residing in the liver, which secrete immunosuppressive molecules, such as vascular endothelial growth factor (VEGF), TGF-β and arginase that suppress T-cells activation [9], a strong immunosuppressive milieu is created, leading to immune escape and enabling uncontrolled tumour growth (Figure 1).
2.3. The Immune Checkpoints, CTLA-4 and PD-1
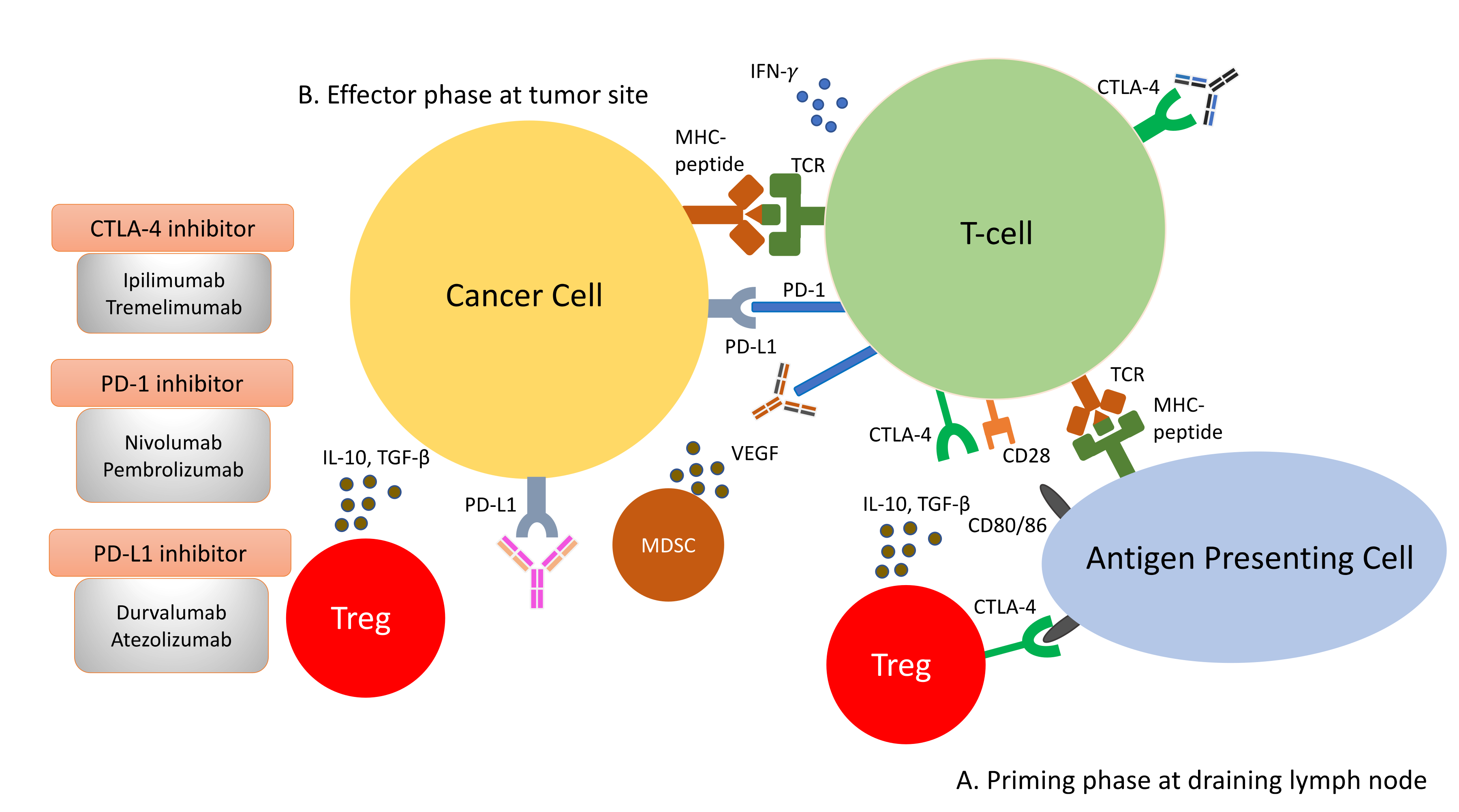
3. The Paradox of Immune Checkpoint Inhibitors and NASH-HCC
3.1. Are NASH-HCC Poor Responders to ICI?
While the extraordinary ORR and OS offered by ICI have been heralded as the game changer in the management of advanced HCC, a recent paper published in Nature titled ‘NASH Limits Antitumour Surveillance in Immunotherapy’, has provided shocking evidence of the potential detrimental effect of ICI in NASH-HCC [20]. The study first demonstrated that the frequency of CD8+PD1+ T-cells was specifically increased in NASH-HCC in mice models. Because of these high numbers of T-cells in NASH-HCC, it was initially thought that the anti-PD-1 treatment may serve as an effective treatment. Surprisingly, when NASH-HCC mice were treated with anti-PD-1 therapy, none of the tumour regressed. Instead, there was an increased incidence of liver fibrosis and liver cancer. However, in non-NASH-HCC mice, a shrinkage of tumours was observed after anti-PD-1 therapy. These findings suggested that anti-PD-1 therapy failed to reinvigorate CT8+PD1+ T-cells to perform effective immunosurveillance. Instead, anti-PD-1 therapy promoted tissue damage and malignant changes. To further verify the function of CD8+PD1+ T-cells, the group depleted NASH-mice of CD8+PD1+ T-cells and found a significant drop in the incidence of HCC in these mice. Treating anti-PD-1 therapy prophylactically in NASH-mice increased CD8+PD1+ T-cells, aggravated liver damage and heightened the incidence of HCC. This evidence provided strong support of the carcinogenic role of CD8+PD1+ T-cells in NASH-mice. The group took their study further to examine if similar findings could be extrapolated to human-NASH. Using single-cell RNA sequencing, they demonstrated that the CD8+PD1+ T-cells in patients with NASH shared similar gene expression patterns with those in NASH-mice. Subsequently, the group conducted a meta-analysis of three published phase III trials (CheckMate-459, KEYNOTE-240 and IMbrave150) comprising a total of 1656 patients to study the effect of immunotherapy in HCC according to the underlying aetiologies. The meta-analysis revealed that immunotherapy did not improve OS in non-viral HCC (HR: 0.92, 95%; CI: 0.77–1.11), whereas those with viral HCC derived survival benefits from immunotherapy (HR: 0.64, 95%; CI: 0.48–0.94). To isolate the effect of anti-PD-L1 immunotherapy with respect to the underlying aetiology of liver damage, the group investigated two additional small cohorts of NAFLD-HCC patients. Consistent with their hypothesis, NAFLD-HCC patients had a shortened OS after immunotherapy, as compared to patients with other aetiologies. In the first validation cohort (n = 130), the median OS was 5.4 months for NAFLD-HCC vs. 11.0 months for HCC of non-NAFLD aetiology; in the second validation cohort (n = 118), the median OS was 8.8 months vs. 17.7 months. Collectively, this study signalled a proposition that immunotherapy might not confer beneficial effects in NAFLD/NASH-HCC.3.2. Progression from NASH to NASH-HCC
In the past few years, new knowledge has been added to our understanding on the molecular mechanisms that drive the transition from NAFLD/NASH to NASH-HCC. These findings support a parallel and multiple-hits paradigm of immune cell-hepatocyte interactions that contribute to the development NASH-HCC [21][22][23]. Firstly, CD8+ T-cells and NKT-cells were demonstrated to cooperatively induce liver damage and carcinogenesis via interaction with hepatocytes in a NASH-mouse model [23]. In addition, the metabolic activation of intrahepatic NKT-cells caused steatosis via the secretion of LIGHT. These interactions resulted in the downregulation of metabolic machinery in hepatocytes, thereby increasing oxidative stress and the activation of procarcinogenic pathways, such as LTβR and canonical NF-κB signalling, promoting a NASH-to-HCC transition [23]. Secondly, platelet aggregations as the initial inflammatory process, in the context of steatosis, were demonstrated to drive NASH to the NASH-HCC transition [24]. This phenomenon was mediated via the interactions between the platelet-specific glycoprotein Ib-α (GPIbα) with Kupffer cells. Antiplatelet therapy was demonstrated to reduce intrahepatic platelet accumulation and the frequency of platelet–immune cell interaction, attenuating NASH and NASH-related HCC in the mouse model [24]. Indeed, a recently published meta-analysis (n = 2,389,019) demonstrated that the use of aspirin was associated with significantly lower HCC risk (RR: 0.61, 95%; CI: 0.51–0.73) [25]. Thirdly, it was noted that in NASH-mice or patients with NASH, there was a distinctive group of residential CD8+ T-cells in the liver that demonstrated an auto-aggressive killing of cells in an MHC-class-I independent fashion [26]. Mechanistically, IL-15 induced FOXO-1 downregulation and CXCR6 upregulation, which together rendered liver-resident CXCR6+CD8+PD1+ T-cells susceptible to metabolic stimuli, such as the acetate released by hepatocytes with steatosis. These CXCR6+CD8+ T-cells then secreted tumour necrosis factor (TNF) and injured hepatocytes. In addition, these activated CXCR6+CD8+PD1+ T-cells caused hepatocyte cell death through a Fas ligand-dependent apoptosis. This auto-aggressive mechanism of cell killing mediated via CXCR6+CD8+PD1+ T-cells was fundamentally distinct from that by antigen presentation in protective adaptive immunity. It has been postulated that this auto-aggression of CXCR6+CD8+PD1+ T-cells in the liver is responsible for the chronic liver damage in NASH and may be implicated in the development of HCC (Figure 1).3.3. Unresolved Questions in NASH-HCC and ICI
The meta-analysis conducted by Pfister et al. raised some important key questions [20]. Contrary to the preclinical findings of enhanced tumour progression in NASH-mice treated with ICI, the ORRs of non-viral HCC patients receiving nivolumab or pembrolizumab appeared similar to patients with viral HCC in the range of 20% [27] (Table 1). Subgroup analyses for patients treated with atezolizumab and bevacizumab demonstrated similar improvement in PFS, regardless of the underlying causes of HCC [28][29]. The favourable ORRs and PFS in the non-viral HCC patients without improvement in OS have led to several alternative hypotheses. Firstly, there was a fundamental limitation of the meta-analysis, in which it included a heterogeneous population of non-viral HCC patients. Non-viral HCC includes a wide spectrum of aetiologies, such as NASH, alcoholic steatohepatitis, cryptogenic cirrhosis, primary biliary cirrhosis, and autoimmune hepatitis, etc. A lack of stratification of the underlying liver disease would make it difficult to tease out the effect of ICI on NASH-HCC. Secondly, concluding ICI was ineffective in non-viral HCC, based solely on the HR without looking at the comparator arm, is inappropriate. The control arm of the two included trials (IMbrave150 and CheckMate-459) in the meta-analysis was sorafenib. The HRs demonstrated that, relative to sorafenib, ICI demonstrated OS benefits in viral HCC but not in non-viral HCC (viral HCC—HR: 0.64 vs. non-viral HCC—HR: 0.92) [30]. It is commonly thought that non-viral HCC patients in general respond better with molecular-targeted agents (e.g., sorafenib) because they often present with favourable clinical–pathological characteristics, such as the absence of cirrhosis [31][32][33]. In particular, non-viral HCC patients treated with sorafenib demonstrated a much longer median OS compared to viral HCC in the IMbrave150 trial [28]. Therefore, the more appropriate postulation would be that the longer survival benefits of sorafenib in non-viral HCC diluted the benefits of ICI, resulting in a seeming lack of benefits of ICI in non-viral HCC. Alternatively, it could be that the survival benefits of ICI are much more pronounced in viral HCC, as observed in the Asian populations, where viral HCC is predominant [34]. Thirdly, it is uncertain whether there were differences in the access to downstream treatments in patients with non-viral HCC in the control group, which could have driven better outcomes. Fourthly, the sequence of treatments might have impacts on the outcome in patients with non-viral HCC, as observed in the management of other malignancies. In a small retrospective study evaluating 25 patients with BRAF-mutant melanoma, the treatment with immunotherapy after a BRAF inhibitor resulted in higher ORR, compared to giving immunotherapy before a BRAF inhibitor (43.8% vs. 0%). The OS, however, was not different [35]. To answer these questions, well-designed clinical trials will be needed to draw definite conclusions on the effectiveness of ICI in NASH-HCC.| Study | Aetiology (Proportion) | Tx | Control | ORR (%) (Tx Arm) | ORR (%) (Control Arm) |
|---|---|---|---|---|---|
| IMbrave150 [28] | HBV (48%) | Atezolizumab + Bevacizumab | Sorafenib | 32 | 8 |
| IMbrave150 [28] | HCV (21%) | Atezolizumab + Bevacizumab | Sorafenib | 30 | 21 |
| IMbrave150 [28] | Non-viral (31%) | Atezolizumab + Bevacizumab | Sorafenib | 27 | 9 |
| CheckMate 459 [36] | HBV (31%) | Nivolumab | Sorafenib | 19 | 8 |
| CheckMate 459 [36] | HCV (23%) | Nivolumab | Sorafenib | 17 | 7 |
| CheckMate 459 [36] | Non-viral (45%) | Nivolumab | Sorafenib | 12 | 7 |
| KEYNOTE-224 [37] | HBV (21%) | Pembrolizumab | - | 24 | - |
| KEYNOTE-224 [37] | HCV (25%) | Pembrolizumab | - | 8 | - |
| KEYNOTE-224 [37] | Non-viral (55%) | Pembrolizumab | - | 30 | - |
| CheckMate 040 (dose expansion) [38] | HBV (24%) | Nivolumab | - | 14 | - |
| CheckMate 040 (dose expansion) [38] | HCV (23%) | Nivolumab | - | 20 | - |
| CheckMate 040 (dose expansion) [38] | Non-viral * (53%) | Nivolumab | - | 22 | - |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers. 2021, 7, 6.
- Ribatti, D. The concept of immune surveillance against tumors. The first theories. Oncotarget 2017, 8, 7175–7180.
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668.
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570.
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998.
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006.
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell Mol. Immunol. 2016, 13, 267–276.
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232.
- Cancer Genome Atlas Research Network. Electronic address wbe Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23.
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16.
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172.
- Deng, L.; He, K.; Pan, Y.; Wang, H.; Luo, Y.; Xia, Q. The role of tumor-associated macrophages in primary hepatocellular carcinoma and its related targeting therapy. Int. J. Med. Sci. 2021, 18, 2109–2116.
- Chan, L.L.; Chan, S.L. Emerging immune checkpoint inhibitors for the treatment of hepatocellular carcinoma. Expert Opin. Emerg. Drugs 2021, 26, 39–52.
- Dong, Y.; Wong, J.S.L.; Sugimura, R.; Lam, K.O.; Li, B.; Kwok, G.G.W.; Leung, R.; Chiu, J.W.Y.; Cheung, T.T.; Yau, T. Recent Advances and Future Prospects in Immune Checkpoint (ICI)-Based Combination Therapy for Advanced HCC. Cancers 2021, 13, 1949.
- Kudo, M. Scientific Rationale for Combination Immunotherapy of Hepatocellular Carcinoma with Anti-PD-1/PD-L1 and Anti-CTLA-4 Antibodies. Liver Cancer 2019, 8, 413–426.
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787.
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The P.D1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550.
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217.
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456.
- Michelotti, A.; de Scordilli, M.; Palmero, L.; Guardascione, M.; Masala, M.; Roncato, R.; Foltran, L.; Ongaro, E.; Puglisi, F. NAFLD-Related Hepatocarcinoma: The Malignant Side of Metabolic Syndrome. Cells 2021, 10, 2034.
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428.
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic C.D8+ T cells and N.KT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564.
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet G.PIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655.
- Memel, Z.N.; Arvind, A.; Moninuola, O.; Philpotts, L.; Chung, R.T.; Corey, K.E.; Simon, T.G. Aspirin Use Is Associated with a Reduced Incidence of Hepatocellular Carcinoma: A Systematic Review and Meta-analysis. Hepatol. Commun. 2021, 5, 133–143.
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Huser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive C.XCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449.
- Kelley, R.K.; Greten, T.F. Hepatocellular Carcinoma—Origins and Outcomes. N. Engl. J. Med. 2021, 385, 280–282.
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from I.Mbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2021, 76, 862–873.
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905.
- Kudo, M. Impaired Response to Immunotherapy in Non-Alcoholic Steatohepatitis-Related Hepatocellular Carcinoma? Liver Cancer 2021, 10, 289–295.
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117.
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1.
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S.; Group H-NIS. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology 2016, 63, 827–838.
- Kudo, M.; Lim, H.Y.; Cheng, A.L.; Chao, Y.; Yau, T.; Ogasawara, S.; Kurosaki, M.; Morimoto, N.; Ohkawa, K.; Yamashita, T.; et al. Pembrolizumab as Second-Line Therapy for Advanced Hepatocellular Carcinoma: A Subgroup Analysis of Asian Patients in the Phase 3 KEYNOTE-240 Trial. Liver Cancer 2021, 10, 275–284.
- Aya, F.; Fernandez-Martinez, A.; Gaba, L.; Victoria, I.; Tosca, M.; Pineda, E.; Gascon, P.; Prat, A.; Arance, A. Sequential treatment with immunotherapy and B.RAF inhibitors in B.RAF-mutant advanced melanoma. Clin. Transl. Oncol. 2017, 19, 119–124.
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2021, 23, 77–90.
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952.
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502.