Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Dipak Banerjee and Version 3 by Camila Xu.
Tunicamycin, is a small biological when tested in Matrigel™ implants for 10 days in athymic nude [Balb/c (nu/nu)] moleicule, that blocks a specific stepe, macroscopic as well as H&E-stained microscopic images of the protein N-glycosylation pathway in the endoplasmic reticulum (ER), Matrigel™ sections indicate the absence of microvessels (i.e., the catalytic activity of N-acetylglusosaminyl 1-phosphate transferase (GPTblood capillaries).
- tunicamycin
- N-acetylglucosaminyl 1-phosphate transferase
- ER stress
- unfolded protein response
- protein N-glycosylation inhibitor
- asparagine-linked protein glycosylation
1. Effect of Tunicamycin on In Vivo Angiogenesis
1. Effect of Tunicamycin on In Vivo Angiogenesis
Tunicamycin, when tested in Matrigel™ implants for 10 days in athymic nude [Balb/c (nu/nu)] mice, macroscopic as well as H&E-stained microscopic images of the Matrigel™ sections indicate the absence of microvessels (i.e., blood capillaries). Immunohistochemistry of CD34 and CD144 in Matrigel™ sections also supports their reduced expression and explains the presence of quantitatively few blood vessels in Tunicamycin treated Matrigel™ sections. Enhanced protein and mRNA expression of the endogenous anti-angiogenic factor Thrombospondin-1 (Tsp-1) in Tunicamycin-treated Matrigel™ plugs as well as in Tunicamycin-treated capillary endothelial cells supports inhibition of angiogenesis. The anti-angiogenic effect of Tunicamycin is further supported by the decreased Matrigel™ invasion and chemotaxis of Tunicamycin-treated capillary endothelial cells even when VEGF is present [71].
Tunicamycin, when tested in Matrigel™ implants for 10 days in athymic nude [Balb/c (nu/nu)] mice, macroscopic as well as H&E-stained microscopic images of the Matrigel™ sections indicate the absence of microvessels (i.e., blood capillaries). Immunohistochemistry of CD34 and CD144 in Matrigel™ sections also supports their reduced expression and explains the presence of quantitatively few blood vessels in Tunicamycin treated Matrigel™ sections. Enhanced protein and mRNA expression of the endogenous anti-angiogenic factor Thrombospondin-1 (Tsp-1) in Tunicamycin-treated Matrigel™ plugs as well as in Tunicamycin-treated capillary endothelial cells supports inhibition of angiogenesis. The anti-angiogenic effect of Tunicamycin is further supported by the decreased Matrigel™ invasion and chemotaxis of Tunicamycin-treated capillary endothelial cells even when VEGF is present [1].
2. Effect of Tunicamycin on Breast Tumor Progression
The inability of VEGF to reverse the anti-angiogenic effect of Tunicamycin both in vitro and in vivo allowed testing of Tunicamycin in humanized breast cancers developed in athymic nude [Balb/c (nu/nu)] mice. The research used double-negative (ER
2. Effect of Tunicamycin on Breast Tumor Progression
The inability of VEGF to reverse the anti-angiogenic effect of Tunicamycin both in vitro and in vivo allowed testing of Tunicamycin in humanized breast cancers developed in athymic nude [Balb/c (nu/nu)] mice. The study used double-negative (ER
−
/PR
−
/HER2
+
) and triple-negative (ER
−
/PR
−
/HER2
−) breast cancer pre-clinical mouse models with tumors developed either orthotopically (double-negative, MDA-MB-435) or as xenograft (triple-negative, MDA-MB-231). Mice with double-negative tumor (orthotopic) have received Tunicamycin through intravenous injection (iv) 0.0–1.0 mg/Kg body weight once a week for three weeks, and the mice with triple-negative tumor (xenograft) received Tunicamycin 0.0–0.25 mg/Kg orally twice a week for three weeks. Control mice received the vehicle only. The double-negative tumor (grade III breast adenocarcinoma), when treated with 1 mg/Kg of Tunicamycin, regressed approximately 55% after three weeks, whereas the triple-negative tumor regressed approximately 65% after one week of 0.25 mg/Kg Tunicamycin treatment given orally [1]. These doses are far lower than the FDA-approved dose for the breast cancer drug Taxol. Taxol requires 15–60 times more to match a comparable effect of Tunicamycin. In the control group, the tumor growth is almost doubled in three weeks.
To evaluate N-glycan status in breast tumors, the tissue sections from the double-negative breast tumor are stained with Texas-Red-WGA conjugate and examined under a fluorescence microscope. The N-glycans in tumor microvessels in untreated controls are stained markedly, but the staining intensity per vessel is reduced almost 50% in Tunicamycin-treated tumors. Tumor cells from untreated control also exhibit positive WGA staining, but the intensity is much reduced in the tumor treated with Tunicamycin. Examination of the paraffin section from the excised tumor after 23 days of Tunicamycin treatment by H&E staining indicates a reduction in microvascular density as the Tunicamycin concentrations are increased from zero to 1 mg/Kg. The mitotic index of tumor cells also declined as a consequence of Tunicamycin treatment. To correlate the tumor growth with cellular markers, the expression of Ki-67 (a cellular proliferation marker) and VEGF (a pro-angiogenic molecule) was analyzed immunohistochemically. The results explain that the expression of both Ki-67 and VEGF in tumors from Tunicamycin-treated mice (1.0 mg/Kg) is reduced significantly. This parallels the reduction of microvessel count and the mitotic index, respectively. The mice expressed no behavioral or skeletal toxicity [1]. Accumulating evidence thus unequivocally supports that Tunicamycin could lead the way to developing a dual-action glycotherapy for treating breast cancer in the clinic. It is anti-angiogenic on one hand and anti-tumorigenic on the other hand. Hence, the effect is holistic.
Inhibition of N-glycan biosynthesis with Tunicamycin has been found to develop “ER stress” and consequently upr-mediated apoptosis [2]. To evaluate whether ER stress also exists in breast tumors when treated with Tunicamycin, both tumor microvasculature and the tumor cells in formalin-fixed paraffin-embedded breast tumor tissue sections were examined. The endothelial cells in tumor microvasculature are identified by staining with anti-CD144 (a marker for endothelial cells) antibody and then stained for the GRP78 (an ER chaperone and the ER stress marker). CD144 and GRP78 co-localize on endothelial cells. CD144-staining of endothelium appears a thin line around the vessel as is the GRP78 in the untreated control. On the other hand, in Tunicamycin-treated tumors, a high expression of GRP78 was observed in tumor microvessels. To understand that it is not an indirect effect because of nutritional deprivation due to reduced blood flow in the tumor, the effect of Tunicamycin is currently being evaluated in multiple subtypes of human breast cancer cells. The preliminary results are encouraging and indicate Tunicamycin inhibits their proliferation (Zhang Z et al.—manuscript under preparation). Thus, from both in vitro and in vivo studies, researchers conclude that Tunicamycin is not only a novel dual-action glycotherapy for treating breast cancer, but also a therapy that may not discriminate between breast cancer subtypes [1][3].
3. Nanoformulation Enhances the Anti-Angiogenic Efficacy of Tunicamycin
Nanoparticles <100 nm evade the immune system’s clearing mechanism long enough to reach the targeted disease tissue efficiently. To evaluate the efficiency and efficacy of nanoformulated Tunicamycin, researchers developed, characterized a series of gold-conjugated Tunicamycin nanoparticles (i.e., Tunicamycin encapsulated in peptide nanotubes, nanotubes bound to gold nanoparticles (Au NPs) conjugated with Tunicamycin, Tunicamycin conjugated with nanotubes, Au NPs bound to tubes and conjugated with Tunicamycin, and Au NPs conjugated with Tunicamycin) and tested them against the in vitro angiogenesis model of capillary endothelial cells in culture. MTT (3-(4,5-methylthiazol-2-yl)-2,5-dipheyl-tetrazolium bromide) assays indicate that nanoparticles (1 μg/mL) inhibit capillary endothelial cells proliferation, ~50% within one hour of treatment, whereas the native Tunicamycin has no effect. The nanoformulated Tunicamycin blocks the cell cycle progression by inhibiting either cyclin D1 and CDK4, or only the cyclin D1, or the CDK4 expression, as well as the expression of phospho Rb (serine-229/threonine-252). Phosphorylation of p53 at serine-392 is down-regulated but not the total p53. Increased expression of GRP-78/Bip identifies “ER stress”. Upregulated expression of phospho-PERK (1.6–5.5-fold) and IRE-1 supports induction of upr. The upr aims to restore protein folding homeostasis at the initial phase, while this signal transduction pathway triggers apoptosis if ER stress remains unmitigated. In mammalian cells, the upr is mediated by three classes of signaling components: PKR-like ER protein kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1). Among the three branches, IRE1 is the most evolutionarily conserved arm from yeast to human. IRE1 has two isoforms: IRE1α and IRE1β; IRE1α is widely expressed in most cells and tissues, while IRE1β is restricted to intestinal epithelial cells. IRE1α is a 100 kDa type I ER-membrane-resident protein consisting of a luminal domain, a transmembrane domain, and a cytoplasmic region. The cytoplasmic region contains a kinase domain and an endoribonuclease (RNase) domain, making IRE1α a bifunctional enzyme. The luminal domain of IRE1α acts as a sensor of the ER unfolded protein load. Combining with the ER chaperone binding immunoglobulin-protein (Bip, also termed GRP78), the luminal domain stays in an inactive monomeric state. The activation of this luminal fragment depends on the dissociation with Bip, rather than direct interaction with unfolded proteins. As for the transmembrane domain, very little is known about its functional and structural roles thus far. The kinase domain of IRE1α serves as a substrate for IRE1α trans-autophosphorylation, and provides an ATP-binding pocket. Autophosphorylation of the kinase domain and binding of ADP (or ATP in vivo) allosterically regulate dimerization/oligomerization and lead to IRE1α RNase activation. IRE1α is the most sensitive of the three upr branches that are triggered to cope with ER stress in mammalian cells. IRE1α signaling is quite a context-specific on account of many adaptor and modulator proteins that directly interact with it, including heat shock proteins (HSPs), RING finger protein 13 (RNF13), poly (ADP-ribose) polymerase 16 (PARP16/ARTD15), Bax/Bak, and Bax inhibitor-1 (BI-1). The activated IRE1α triggers different downstream pathways depending on the UPRosome formed by distinct modulator proteins. At the initial phase of ER stress, the IRE1α-XBP1 axis functions as an adaptive response. While ER stress sustains or intensifies, signals shift to apoptotic responses. Furthermore, IRE1α signaling can be exploited to the development of a wide range of prevalent human diseases, with cancer being the most characterized. PARPs were found to regulate DNA damage repair and the cytoplasmic stress response [4]. Human PARP16 (also known as ARTD15) is a tail-anchored ER transmembrane protein with a cytosolic catalytic domain. The luminal C-tail of PARP16 is required for its function, indicating that stress signals may be transduced from the ER domain to the cytosolic domain [5]. During ER stress, PARP16 enzymatic activity is upregulated. Co-immunoprecipitation assay demonstrated a robust association between PARP16 and IRE1α both in the presence and absence of ER stress. The ADP-ribosylation caused by PARP16 increases the kinase and RNase activities of IRE1α. Under ER stress, PARP16 could facilitate Bip dissociation from IRE1α. This effect was impaired in PARP16 knockdown, accompanied by an increase of cell death, suggesting PARP16 is involved in cell survival [6]. Down-regulated expression of caspase-9 and caspase-3 suggests a non-canonical pathway of cell death by nanoformulated Tunicamycin [7]. Thus, the take-home message is that nanoformulated Tunicamycin prevents capillary proliferation, which in turn reduces the nutrient flow to the tumor and consequently causes their death by starvation.4. Tunicamycin Interferes with Wnt Signaling and Inhibits Angiogenesis
Wnts are a family of secreted growth factors involved in cellular differentiation to organogenesis, and have been suggested to function in angiogenesis. Chemically, they are glycoproteins whose accumulation in the extracellular matrix activates pathways in adjacent cells. Wnt ligands bind to Frizzled receptor, a member of a family of seven-pass transmembrane proteins [8]. A co-receptor, LRP5/6 is required to activate the Wnt/β-catenin and Planar Cell Polarity (PCP) pathways which can be blocked when LRP5/6 is bound to a secreted regulatory protein, Dickkopf-1. Frizzled is a G protein-coupled receptor [9], and Wnt binding to frizzled can activate more than one distinct branch of the Wnt signaling cascade. The different branches of the Wnt signaling pathway are designated the “canonical” signaling pathway, which is referred to as the Wnt/β-catenin signaling pathway, and the “non-canonical”, which includes Wnt signaling through Ca) breast cancer pre-clinical mouse models with tumors developed either orthotopically (double-negative, MDA-MB-435) or as xenograft (triple-negative, MDA-MB-231). Mice with double-negative tumor (orthotopic) have received Tunicamycin through intravenous injection (iv) 0.0–1.0 mg/Kg body weight once a week for three weeks, and the mice with triple-negative tumor (xenograft) received Tunicamycin 0.0–0.25 mg/Kg orally twice a week for three weeks. Control mice received the vehicle only. The double-negative tumor (grade III breast adenocarcinoma), when treated with 1 mg/Kg of Tunicamycin, regressed approximately 55% after three weeks, whereas the triple-negative tumor regressed approximately 65% after one week of 0.25 mg/Kg Tunicamycin treatment given orally [71]. These doses are far lower than the FDA-approved dose for the breast cancer drug Taxol. Taxol requires 15–60 times more to match a comparable effect of Tunicamycin. In the control group, the tumor growth is almost doubled in three weeks.
To evaluate N-glycan status in breast tumors, the tissue sections from the double-negative breast tumor are stained with Texas-Red-WGA conjugate and examined under a fluorescence microscope. The N-glycans in tumor microvessels in untreated controls are stained markedly, but the staining intensity per vessel is reduced almost 50% in Tunicamycin-treated tumors. Tumor cells from untreated control also exhibit positive WGA staining, but the intensity is much reduced in the tumor treated with Tunicamycin. Examination of the paraffin section from the excised tumor after 23 days of Tunicamycin treatment by H&E staining indicates a reduction in microvascular density as the Tunicamycin concentrations are increased from zero to 1 mg/Kg. The mitotic index of tumor cells also declined as a consequence of Tunicamycin treatment. To correlate the tumor growth with cellular markers, the expression of Ki-67 (a cellular proliferation marker) and VEGF (a pro-angiogenic molecule) was analyzed immunohistochemically. The results explain that the expression of both Ki-67 and VEGF in tumors from Tunicamycin-treated mice (1.0 mg/Kkg) is reduced significantly. This parallels the reduction of microvessel count and the mitotic index, respectively. The mice expressed no behavioral or skeletal toxicity [71]. Accumulating evidence thus unequivocally supports that Tunicamycin could lead the way to developing a dual-action glycotherapy for treating breast cancer in the clinic. It is anti-angiogenic on one hand and anti-tumorigenic on the other hand. Hence, the effect is holistic.
Inhibition of N-glycan biosynthesis with Tunicamycin has been found to develop “ER stress” and consequently upr-mediated apoptosis [72]. To evaluate whether ER stress also exists in breast tumors when treated with Tunicamycin, both tumor microvasculature and the tumor cells in formalin-fixed paraffin-embedded breast tumor tissue sections were examined. The endothelial cells in tumor microvasculature are identified by staining with anti-CD144 (a marker for endothelial cells) antibody and then stained for the GRP78 (an ER chaperone and the ER stress marker). CD144 and GRP78 co-localize on endothelial cells. CD144-staining of endothelium appears a thin line around the vessel as is the GRP78 in the untreated control. On the other hand, in Tunicamycin-treated tumors, a high expression of GRP78 was observed in tumor microvessels. To understand that it is not an indirect effect because of nutritional deprivation due to reduced blood flow in the tumor, the effect of Tunicamycin is currently being evaluated in multiple subtypes of human breast cancer cells. The preliminary results are encouraging and indicate Tunicamycin inhibits their proliferation (Zhang Z et al.—manuscript under preparation). Thus, from both in vitro and in vivo studies, we conclude that Tunicamycin is not only a novel dual-action glycotherapy for treating breast cancer, but also a therapy that does may not discriminate between breast cancer subtypes [71,73].
3. Nanoformulation Enhances the Anti-Angiogenic Efficacy of Tunicamycin
Nanoparticles <100 nm evade the immune system’s clearing mechanism long enough to reach the targeted disease tissue efficiently. To evaluate the efficiency and efficacy of nanoformulated Tunicamycin, we developed, characterized a series of gold-conjugated Tunicamycin nanoparticles (i.e., Tunicamycin encapsulated in peptide nanotubes, nanotubes bound to gold nanoparticles (Au NPs) conjugated with Tunicamycin, Tunicamycin conjugated with nanotubes, Au NPs bound to tubes and conjugated with Tunicamycin, and Au NPs conjugated with Tunicamycin) and tested them against the in vitro angiogenesis model of capillary endothelial cells in culture. MTT (3-(4,5-methylthiazol-2-yl)-2,5-dipheyl-tetrazolium bromide) assays indicate that nanoparticles (1 μg/mL) inhibit capillary endothelial cells proliferation, ~50% within one hour of treatment, whereas the native Tunicamycin has no effect. The nanoformulated Tunicamycin blocks the cell cycle progression by inhibiting either cyclin D1 and CDK4, or only the cyclin D1, or the CDK4 expression, as well as the expression of phospho Rb (serine-229/threonine-252). Phosphorylation of p53 at serine-392 is down-regulated but not the total p53. Increased expression of GRP-78/Bip identifies “ER stress”. Upregulated expression of phospho-PERK (1.6–5.5-fold) and IRE-1 supports induction of upr.
The upr aims to restore protein folding homeostasis at the initial phase, while this signal transduction pathway triggers apoptosis if ER stress remains unmitigated. In mammalian cells, the upr is mediated by three classes of signaling components: PKR-like ER protein kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1). Among the three branches, IRE1 is the most evolutionarily conserved arm from yeast to human. IRE1 has two isoforms: IRE1α and IRE1β; IRE1α is widely expressed in most cells and tissues, while IRE1β is restricted to intestinal epithelial cells.
IRE1α is a 100 kDa type I ER-membrane-resident protein consisting of a luminal domain, a transmembrane domain, and a cytoplasmic region. The cytoplasmic region contains a kinase domain and an endoribonuclease (RNase) domain, making IRE1α a bifunctional enzyme. The luminal domain of IRE1α acts as a sensor of the ER unfolded protein load. Combining with the ER chaperone binding immunoglobulin-protein (Bip, also termed GRP78), the luminal domain stays in an inactive monomeric state. The activation of this luminal fragment depends on the dissociation with Bip, rather than direct interaction with unfolded proteins. As for the transmembrane domain, very little is known about its functional and structural roles thus far. The kinase domain of IRE1α serves as a substrate for IRE1α trans-autophosphorylation, and provides an ATP-binding pocket. Autophosphorylation of the kinase domain and binding of ADP (or ATP in vivo) allosterically regulate dimerization/oligomerization and lead to IRE1α RNase activation.
IRE1α is the most sensitive of the three upr branches that are triggered to cope with ER stress in mammalian cells. IRE1α signaling is quite a context-specific on account of many adaptor and modulator proteins that directly interact with it, including heat shock proteins (HSPs), RING finger protein 13 (RNF13), poly (ADP-ribose) polymerase 16 (PARP16/ARTD15), Bax/Bak, and Bax inhibitor-1 (BI-1). The activated IRE1α triggers different downstream pathways depending on the UPRosome formed by distinct modulator proteins. At the initial phase of ER stress, the IRE1α-XBP1 axis functions as an adaptive response. While ER stress sustains or intensifies, signals shift to apoptotic responses. Furthermore, IRE1α signaling can be exploited to the development of a wide range of prevalent human diseases, with cancer being the most characterized.
PARPs were found to regulate DNA damage repair and the cytoplasmic stress response [77]. Human PARP16 (also known as ARTD15) is a tail-anchored ER transmembrane protein with a cytosolic catalytic domain. The luminal C-tail of PARP16 is required for its function, indicating that stress signals may be transduced from the ER domain to the cytosolic domain [78]. During ER stress, PARP16 enzymatic activity is upregulated. Co-immunoprecipitation assay demonstrated a robust association between PARP16 and IRE1α both in the presence and absence of ER stress. The ADP-ribosylation caused by PARP16 increases the kinase and RNase activities of IRE1α. Under ER stress, PARP16 could facilitate Bip dissociation from IRE1α. This effect was impaired in PARP16 knockdown, accompanied by an increase of cell death, suggesting PARP16 is involved in cell survival [79].
Down-regulated expression of caspase-9 and caspase-3 suggests a non-canonical pathway of cell death by nanoformulated Tunicamycin [80]. Thus, the take-home message is that nanoformulated Tunicamycin prevents capillary proliferation, which in turn reduces the nutrient flow to the tumor and consequently causes their death by starvation.
4. Tunicamycin Interferes with Wnt Signaling and Inhibits Angiogenesis
Wnts are a family of secreted growth factors involved in cellular differentiation to organogenesis, and have been suggested to function in angiogenesis. Chemically, they are glycoproteins whose accumulation in the extracellular matrix activates pathways in adjacent cells. Wnt ligands bind to Frizzled receptor, a member of a family of seven-pass transmembrane proteins [81]. A co-receptor, LRP5/6 is required to activate the Wnt/β-catenin and Planar Cell Polarity (PCP) pathways which can be blocked when LRP5/6 is bound to a secreted regulatory protein, Dickkopf-1. Frizzled is a G protein-coupled receptor [82], and Wnt binding to frizzled can activate more than one distinct branch of the Wnt signaling cascade. The different branches of the Wnt signaling pathway are designated the “canonical” signaling pathway, which is referred to as the Wnt/β-catenin signaling pathway, and the “non-canonical”, which includes Wnt signaling through Ca
2+, PCP and other signaling mechanisms that do not involve β-catenin. In canonical Wnt signaling, the level of cytosolic β-catenin is transiently activated by activating Disheveled (Dvl) to block the phosphorylation of β-catenin by GSK-3. Stabilization of β-catenin promotes its nuclear translocation where it interacts with a family of transcription factors Lef/Tcf [10], activating Wnt/β-catenin target genes, including vascular endothelial growth factor A (VEGF-A), a stimulator of angiogenesis. Therefore, it is ideal to evaluate whether the Wnt pathway is affected in Tunicamycin-treated capillary endothelial cells. The results are expected to provide insight into whether a canonical or a non-canonical pathway is active under the experimental conditions.
, PCP and other signaling mechanisms that do not involve β-catenin. In canonical Wnt signaling, the level of cytosolic β-catenin is transiently activated by activating Disheveled (Dvl) to block the phosphorylation of β-catenin by GSK-3. Stabilization of β-catenin promotes its nuclear translocation where it interacts with a family of transcription factors Lef/Tcf [83], activating Wnt/β-catenin target genes, including vascular endothelial growth factor A (VEGF-A), a stimulator of angiogenesis. Therefore, it is ideal to evaluate whether the Wnt pathway is affected in Tunicamycin-treated capillary endothelial cells. The results are expected to provide insight into whether a canonical or a non-canonical pathway is active under the experimental conditions. We used immunofluorescence microscopy and Western blotting as tools, and detected the presence of Frizzled, APC, and β-catenin in the capillary endothelial cell model. Treating cells for 32 h with Tunicamycin causes downregulation of β-catenin expression (Figure 4 and Figure 5). The results thus support that Tunicamycin interferes with the canonical branch of the wnt signaling and impacts angiogenesis.
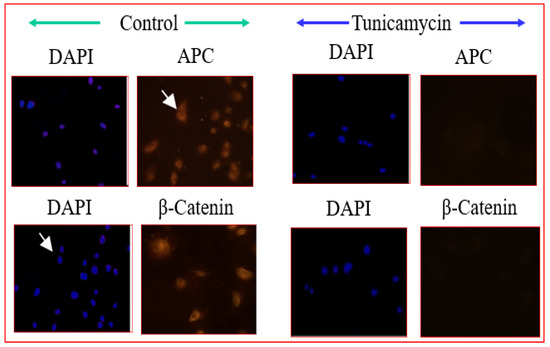
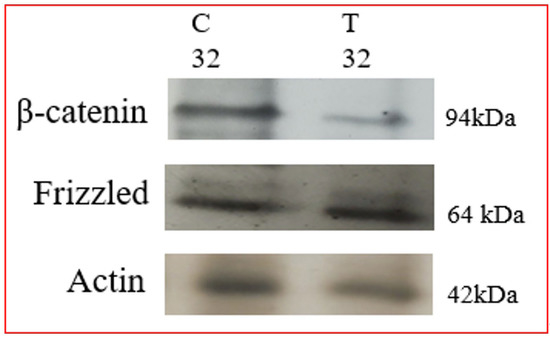
Figure 4. Immunofluorescence microscopy of Wnt proteins: The capillary endothelial cells were cultured for 24 h and synchronized for 72 h. The cells were treated with or without Tunicamycin (1 μg/mL) for 32 h, fixed with 90% ice-cold methanol for 30 s, permeabilized with Triton™ X-100 for 10 min, washed with PBS, pH 7.4 3 times, blocked with 3% BSA in PBS, pH 7.4 for 30 min, treated with anti-β-catenin, and anti-APC antibodies (1:50; v/v) overnight followed by Rhodamin-conjugated rabbit anti-mouse IgG (red) secondary antibody (1:100; v/v) before collecting the images in a Zeiss Axioskop 2. The nucleus was stained with Hoechst dye (blue).
Figure 5. Western blotting of Wnt proteins: The capillary endothelial cells were cultured for 24 h and synchronized for 72 h. The cells were treated with or without Tunicamycin (1 μg/mL) for 32 h. At the end, the proteins were separated on 10% SDS-PAGE and transferred to nitrocellulose membranes. The blots were treated with anti-β-catenin and anti-frizzled monospecific antibodies followed by HRP-conjugated secondary antibodies and developed with EC reagents.