Estrogen receptor (ER) signalling promotes proliferation in breast cancer (BCa). Endocrine therapies (ET) target this signaling by preventing the production of its ligand estrogen, or by blocking its interaction with the ER. The ER is regulated by a range of posttranslational modifications (PTMs) including ubiquitylation, SUMOylation, phosphorylation, palmitoylation, acetylation, methylation and glycosylation. These PTMs control ER activity, stability as well as its interactions with other proteins. For example, kinases play significant roles in mediating crosstalk between the ER and the PI3K-AKT-mTOR and MAPK pathways, which are implicated in ET resistance. Another major control of ER activity is through the regulation of its half-life, largely by ubiquitin which initiates degradation through the ubiquitin proteasome system. As such, PTMs play a central role in the regulation of ER activity and stability.
- Breast Cancer
- Estrogen Receptor
- Posttranslational Modification
- Stability
- Endocrine Therapy
1. Introduction
The estrogen receptor (ER) is the main downstream effector of its ligand estrogen and has functions connected to the menstrual cycle, pregnancy, and lactation in females and in maintaining cardiovascular, nervous, musculoskeletal, and immune system functioning [1]. There are two subtypes of ER, namely ERα and ERβ, encoded by ESR1 and ESR2, respectively. These genes are located on separate chromosomes, ESR1 at position 6q24-27 and ESR2 at 14q22-24 [2,3,4]. Whilst highly similar, their ligand binding domains (LBD) differ, enabling specific physiological functions [1,5,6].
The ERα, a 66 kDa protein, is comprised of four domains, namely activation function 1 (AF-1), DNA binding domain (DBD), the hinge region, and the LBD also known as AF-2 [1,7] (Figure 1).
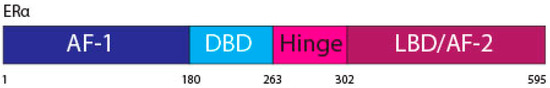
Figure 1. ERα functional domains. The ERα functional domains include; Activation Function 1 (AF-1) (purple), DNA Binding Domain (DBD) (blue), hinge (pink), Ligand Binding Domain (LBD) and AF-2 (plum).
Upon estrogen binding, ER forms homodimers (ERα/ERα or ERβ/ERβ) and heterodimers (ERα/ERβ) [8,9]. This review focuses on ESR1 encoding ERα, referred to as ER henceforth. Following dimerisation, ER translocates to the nucleus and regulates transcription through estrogen response element (ERE) binding within target gene promoters (Figure 2, “genomic function”).
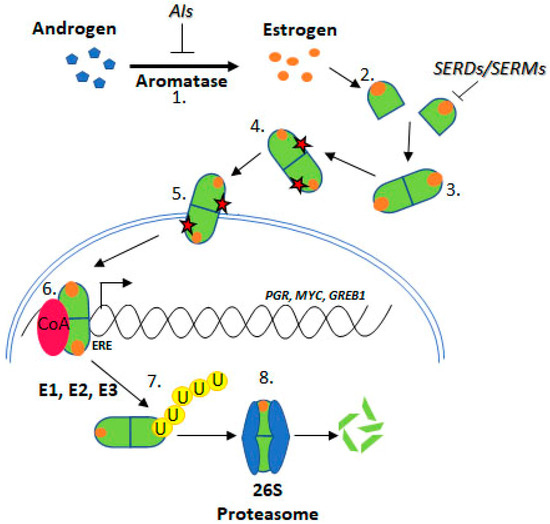
Figure 2. Estrogen Receptor Signalling and ER Targeting Treatment. Aromatase converts androgens to estrogens, Estrogen receptor binds estrogen, Dimerisation, Post translational modification, Nuclear localisation, Coactivator binding and target gene (PGR, c-Myc, GREB1) transcription, Addition of ubiquitin by E1, E2 & E3 ligases, Degradation by the 26S proteasome. Aromatase Inhibitors (AIs) inhibit the enzyme aromatase, Selective Estrogen Receptor Degraders (SERDs) and Selective Estrogen Receptor Modifiers (SERMs) prevent estrogen binding.
Alternatively, ER may crosstalk with the PI3K-AKT-mTOR or MAPK pathways, in a “non-genomic” manner (see below), in both scenarios promoting cell proliferation and suppressing apoptosis [10].
Approximately 75% of BCa express ER, which promotes oncogenesis. As such, the ER is a common target for BCa treatment using endocrine therapies (ETs) including tamoxifen, fulvestrant, and aromatase inhibitors (AIs) [11] (Figure 2). Tamoxifen is known as a selective ER modifier (SERM) and fulvestrant as a selective ER degrader (SERD) [12]. SERMs competitively bind ER forming an inactive complex, by preventing coactivator interactions [12,13,14,15]. SERDs also competitively bind ER; however, their binding targets the receptor for proteasomal degradation [16,17,18]. AIs are a group of drugs that prevent the synthesis of the estrogen, through inhibition of aromatase [19,20,21]. AIs include letrozole, anastrozole, and exemestane. Whilst these treatments are initially effective for many ER positive BCa patients, resistance remains a significant issue.
Resistance to ET is common, approximately 30% of BCa patients acquire resistance [22,23]. ESR1 mutations have been identified in ET resistant BCa tumours. Several ESR1 mutations have been functionally characterised and confer key attributes associated with ET resistance, indicating mechanistic roles in resistance such as estrogen independence, increased transcription of ER target genes like PGR (progesterone receptor), GREB1 (growth regulation by estrogen in breast cancer 1), and MYC (c-myc), and increased proliferation and altered ER conformation [24,25] (Figure 2).
Since a major control of ER activity is through regulation of its half-life, changes to ER stability may influence sensitivity to ET. For example, posttranslational modifications (PTMs) and ESR1 mutations causing amino acid substitutions at PTM sites may influence ER stability, and ultimately activity.
2. Estrogen Receptor Signaling
The estrogen/ER complex can result in activation of two distinct types of signaling pathways, the genomic and non-genomic. In the genomic pathway, the ER regulates gene transcription through direct binding of its DBD to promotors containing an ERE, or through interactions with other transcription factors at promoter regions [9,26]. The non-genomic pathway, however, enables rapid signaling through bidirectional crosstalk with PI3K-AKT-mTOR and MAPK pathways. These pathways are frequently upregulated in BCa and enable signaling in an estrogen independent manner [27,28,29]. Several kinases from these pathways phosphorylate the ER at various sites, mediating ER stability, localization, and transactivational capacity, discussed in detail below [30,31]. Further, these pathways may contribute to ET resistance through phosphorylation at key sites, even in the absence of estrogen [32,33,34,35,36,37]. Additionally, crosstalk between ER and PI3K-AKT-mTOR and MAPK pathways is bidirectional. The ER may activate these pathways through interaction with modulator of non-genomic action of estrogen receptor (MNAR) scaffold protein and subsequent SRC activation [38,39]. Ultimately, these pathways promote cell cycle progression through increased expression of cyclin D1, and suppress apoptosis [38,40]. Understanding these interactions is important as the PI3K and MAPK pathways are frequently active in ET resistant BCa and contribute to ET resistance [27,28,29].
The ER is regulated by a range of PTMs including ubiquitylation, SUMOylation, phosphorylation, palmitoylation, acetylation, methylation and glycosylation [41,42,43,44]. These modifications are proposed to regulate the activity, stability, and interactions of ER with other proteins or DNA, and ESR1 mutations may influence PTMs and hence ER stability and function (Figure 3).
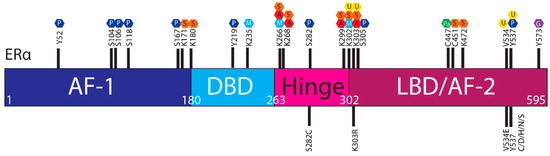
Figure 3. Post translational modifications of ERα. The ERα functional domains include: Activation Function 1 (AF-1) (purple), DNA Binding Domain (DBD) (blue), hinge (pink), Ligand Binding Domain (LBD) and AF-2 (plum). At the top of the schematic are known sites PTMs of ERα; phosphorylation (P, dark blue), SUMOylation (S, orange), methylation (M, light blue), acetylation (A, red), ubiquitylation (U, yellow), pamiltoylation (Pa, green) and gycosylation (G, purple). At the bottom are PTM sites affected by ESR1 mutations.
2.1. Estrogen Receptor Turnover
Whilst ER is produced through transcription and protein synthesis, changes to ER degradation kinetics is the major factor determining ER levels. In BCa, there is an imbalance between the rate of transcription, synthesis and degradation of the ER leading to increased ER stability and thus activity [45]. The half-life of the ER differs significantly depending on estrogen exposure. Generally, the ER half-life is 3–5 h [46] and estrogen presence can reduce the half-life to just 1 h [47]. However, persistent estrogen exposure causes relative ER stability. In fact, in MCF-7 cells exposed to estrogen for 72 h, ER half-life was increased to 6 h, due to the decreased rate of proteolysis associated with p-S118 [48].
2.2. Degradation by the Ubiquitin Proteasome Pathway
Several studies have shown increased ER stability in the presence of the proteasome inhibitor, MG132, indicating that degradation by the ubiquitin–proteasome system (UPS) regulates ER stability [47,49,50,51]. Degradation of the ER occurs predominantly through the UPS, which relies on ER ubiquitylation by ubiquitin activating enzymes (E1) and ubiquitin conjugating enzymes (E2). Conversely, the ER may be protected from degradation through the activity of ubiquitin ligases (E3), which remove ubiquitin from the ER.
Ubiquitin conjugation or removal occurs on lysine residues, and mutations resulting in an exchange, either of such lysines or of amino acids affecting the accessibility of important lysines, may therefore alter the protein’s stability. Additionally, some UPS proteins act as coactivators of nuclear receptors and promote downstream gene transcription [16,44,52,53]. For example, ubiquitin ligase E3A (E6-AP) acts as an ER coactivator and increases gene transcription [16][53]
References
- Cowley, S.M.; Hoare, S.; Mosselman, S.; Parker, M.G. Estrogen Receptors α and β Form Heterodimers on DNA. J. Boil. Chem. 1997, 272, 19858–19862, doi:10.1074/jbc.272.32.19858.
- Kumar, V.; Chambon, P. The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell 1988, 55, 145–156, doi:10.1016/0092-8674(88)90017-7.
- Brown, K.K.; Toker, A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015, 7, 13, doi:10.12703/P7-13.
- Allred, D.C.; Brown, P.; Medina, D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004, 6, 240–245, doi:10.1186/bcr938.
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24, doi:10.1016/j.pharmthera.2017.12.012.
- Jordan, V. Antitumour activity of the antiestrogen ICI 46,474 (Tamoxifen) in the dimethylbenzanthracene (DMBA)—Induced rat mammary carcinoma model. J. Steroid Biochem. 1974, 5, 354, doi:10.1016/0022-4731(74)90388-4.
- Jordan, V.C. Biochemical pharmacology of antiestrogen action. Pharmacol. Rev. 1984, 36.
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937, doi:10.1016/s0092-8674(00)81717-1.
- Casa, A.J.; Hochbaum, D.; Sreekumar, S.; Oesterreich, S.; Lee, A.V. The estrogen receptor alpha nuclear localization sequence is critical for fulvestrant-induced degradation of the receptor. Mol. Cell. Endocrinol. 2015, 415, 76–86, doi:10.1016/j.mce.2015.08.007.
- Dauvois, S.; Danielian, P.S.; White, R.; Parker, M.G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. 1992, 89, 4037–4041, doi:10.1073/pnas.89.9.4037.
- Dauvois, S.; White, R.; Parker, M.G. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J. Cell Sci. 1993, 106, 1377–1388.
- Dowsett, M.; Jones, A.; Johnston, S.R.; Jacobs, S.; Trunet, P.; Smith, I. In vivo measurement of aromatase inhibition by letrozole (CGS 20267) in postmenopausal patients with breast cancer. Clin. Cancer Res. 1995, 1.
- Geisler, J.; King, N.; Anker, G.; Ornati, G.; Di Salle, E.; E Lønning, P.; Dowsett, M. In vivo inhibition of aromatization by exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast cancer patients. Clin. Cancer Res. 1998, 4.
- Geisler, J.; King, N.; Dowsett, M.; Ottestad, L.; Lundgren, S.; Walton, P.; Kormeset, P.; Lonning, P.E. Influence of anastrozole (Arimidex), a selective, non-steroidal aromatase inhibitor, on in vivo aromatisation and plasma oestrogen levels in postmenopausal women with breast cancer. Br. J. Cancer 1996, 74, 1286–1291, doi:10.1038/bjc.1996.531.
- Dowsett, M. Endocrine Resistance in Advanced Breast Cancer. Acta Oncol. 1996, 35, 91–95, doi:10.3109/02841869609083979.
- Haque, M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10, doi:10.3389/fendo.2019.00573.
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.W.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445, doi:10.1038/ng.2822.
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2016, 7, 277–287, doi:10.1158/2159-8290.CD-15-1523.
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170, doi:10.1016/bs.apcsb.2019.01.001.
- Martin, L.-A.; Farmer, I.; Johnston, S.R.D.; Ali, S.; Marshall, C.; Dowsett, M. Enhanced Estrogen Receptor (ER) α, ERBB2, and MAPK Signal Transduction Pathways Operate during the Adaptation of MCF-7 Cells to Long Term Estrogen Deprivation. J. Boil. Chem. 2003, 278, 30458–30468, doi:10.1074/jbc.m305226200.
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224, doi:10.1186/bcr3039.
- Sanchez, C.G.; Ma, C.; Crowder, R.J.; Guintoli, T.; Phommaly, C.; Gao, F.; Lin, L.; Ellis, M.J. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011, 13, R21, doi:10.1186/bcr2833.
- Park, S.; Song, J.; O Joe, C.; Shin, I. Akt stabilizes estrogen receptor α with the concomitant reduction in its transcriptional activity. Cell. Signal. 2008, 20, 1368–1374, doi:10.1016/j.cellsig.2008.03.004.
- Thomas, R.S.; Sarwar, N.; Phoenix, F.; Coombes, R.C.; Ali, S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J. Mol. Endocrinol. 2008, 40, 173-184.
- Barone, I.; Cui, Y.; Herynk, M.H.; Rodriguez, C.-A.; Giordano, C.; Selever, J.; Beyer, A.; Andò, S.; Fuqua, A.W.S. Expression of the K303R estrogen receptor-α breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway. Canc. Res. 2009, 69, 4724-4732.
- Barone, I.; Iacopetta, D.; Covington, K.R.; Cui, Y.; Tsimelzon, A.; Beyer, A.; Andò, S.; Fuqua, S.A.; Anna, T. Phosphorylation of the mutant K303R estrogen receptor α at serine 305 affects aromatase inhibitor sensitivity. Oncogene. 2010, 29, 2404–2414, doi:10.1038/onc.2009.520.
- Holm, C.; Kok, M.; Michalides, R.; Fles, R.; Koornstra, R.; Wesseling, J.; Hauptmann, M.; Neefjes, J.; Peterse, J.; Stål, O.; et al. Phosphorylation of the oestrogen receptor α at serine 305 and prediction of tamoxifen resistance in breast cancer. J. Pathol. 2009, 217, 372–379, doi:10.1002/path.2455.
- Michalides, R.; Griekspoor, A.C.; Balkenende, A.; Verwoerd, D.; Janssen, L.; Jalink, K.; Floore, A.; Velds, A.; VeerL.V.`.; Neefjes, J. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell 2004, 5, 597–605, doi:10.1016/j.ccr.2004.05.016.
- Skliris, G.P.; Nugent, Z.; Watson, P.H.; Murphy, L.C. Estrogen Receptor Alpha Phosphorylated at Tyrosine 537 is Associated with Poor Clinical Outcome in Breast Cancer Patients Treated with Tamoxifen. Horm. Cancer 2010, 1, 215–221, doi:10.1007/s12672-010-0049-z.
- Skliris, G.P.; Nugent, Z.J.; Rowan, B.G.; Penner, C.R.; Watson, P.H.; Murphy, L.C. A phosphorylation code for oestrogen receptor-α predicts clinical outcome to endocrine therapy in breast cancer. Endocr. -Relat. Cancer 2010, 17, 589–597, doi:10.1677/erc-10-0030.
- Cheskis, B.; Greger, J.; Cooch, N.; McNally, C.; McLarney, S.; Lam, H.; Rutledge, S.; Mekonnen, B.; Hauze, D.; Nagpal, S. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids 2008, 73, 901–905, doi:10.1016/j.steroids.2007.12.028.
- Zheng, F.F.; Wu, R.-C.; Smith, C.L.; O’Malley, B.W. Rapid Estrogen-Induced Phosphorylation of the SRC-3 Coactivator Occurs in an Extranuclear Complex Containing Estrogen Receptor. Mol. Cell. Boil. 2005, 25, 8273–8284, doi:10.1128/mcb.25.18.8273-8284.2005.
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; De Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001, 20, 6050–6059, doi:10.1093/emboj/20.21.6050.
- Deng, B.; Tarhan, Y.E.; Ueda, K.; Ren, L.; Katagiri, T.; Park, J.-H.; Nakamura, Y. Critical Role of Estrogen Receptor Alpha O-Glycosylation by N-Acetylgalactosaminyltransferase 6 (GALNT6) in Its Nuclear Localization in Breast Cancer Cells. Neoplasia 2018, 20, 1038–1044, doi:10.1016/j.neo.2018.08.006.
- Faus, H.; Haendler, B. Post-translational modifications of steroid receptors. Biomed. Pharmacother. 2006, 60, 520–528, doi:10.1016/j.biopha.2006.07.082.
- La Rosa, P.; Pesiri, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation Regulates 17β-Estradiol-Induced Estrogen Receptor-α Degradation and Transcriptional Activity. Mol. Endocrinol. 2012, 26, 762–774, doi:10.1210/me.2011-1208.
- Zhang, X.; Tanaka, K.; Yan, J.; Li, J.; Peng, D.; Jiang, Y.; Yang, Z.; Barton, M.C.; Wen, H.; Shi, X. Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc. Natl. Acad. Sci. 2013, 110, 17284–17289, doi:10.1073/pnas.1307959110.
- Tecalco-Cruz, A.C.; O Ramírez-Jarquín, J. Polyubiquitination inhibition of estrogen receptor alpha and its implications in breast cancer. World J. Clin. Oncol. 2018, 9, 60–70, doi:10.5306/wjco.v9.i4.60.
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Boil. 2001, 8, 739–758, doi:10.1016/s1074-5521(01)00056-4.
- Fan, M.; Nakshatri, H.; Nephew, K.P. Inhibiting Proteasomal Proteolysis Sustains Estrogen Receptor-α Activation. Mol. Endocrinol. 2004, 18, 2603–2615, doi:10.1210/me.2004-0164.
- Valley, C.C.; Solodin, N.M.; Powers, G.L.; Ellison, S.J.; Alarid, E.T. Temporal variation in estrogen receptor-α protein turnover in the presence of estrogen. J. Mol. Endocrinol. 2007, 40, 23–34, doi:10.1677/jme-07-0067.
- Kinyamu, H.K.; Collins, J.B.; Grissom, S.F.; Hebbar, P.B.; Archer, T.K. Genome wide transcriptional profiling in breast cancer cells reveals distinct changes in hormone receptor target genes and chromatin modifying enzymes after proteasome inhibition. Mol. Carcinog. 2008, 47, 845–885, doi:10.1002/mc.20440.
- Powers, G.L.; Ellison-Zelski, S.J.; Casa, A.J.; Lee, A.V.; Alarid, E.T. Proteasome inhibition represses ERα gene expression in ER+ cells: A new link between proteasome activity and estrogen signaling in breast cancer. Oncogene 2009, 29, 1509–1518, doi:10.1038/onc.2009.434.
- Powers, G.L.; Rajbhandari, P.; Solodin, N.M.; Bickford, B.; Alarid, E.T. The Proteasome Inhibitor Bortezomib Induces an Inhibitory Chromatin Environment at a Distal Enhancer of the Estrogen Receptor-α Gene. PLoS ONE 2013, 8, e81110, doi:10.1371/journal.pone.0081110.
- Berry, N.B.; Fan, M.; Nephew, K.P. Estrogen receptor-alpha hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol. Endocrinol. 2008, 22, 1535–1551, doi:10.1210/me.2007-0449.
- Tecalco-Cruz, A.C.; Ramirez-Jarquin, J.O.; Cruz-Ramos, E. Estrogen Receptor Alpha and its Ubiquitination in Breast Cancer Cells. Curr. Drug Targets 2019, 20, 690–704, doi:10.2174/1389450119666181015114041.
- Fan, M.; Nakshatri, H.; Nephew, K.P. Inhibiting Proteasomal Proteolysis Sustains Estrogen Receptor-α Activation. Mol. Endocrinol. 2004, 18, 2603–2615, doi:10.1210/me.2004-0164.
- Valley, C.C.; Solodin, N.M.; Powers, G.L.; Ellison, S.J.; Alarid, E.T. Temporal variation in estrogen receptor-α protein turnover in the presence of estrogen. J. Mol. Endocrinol. 2007, 40, 23–34, doi:10.1677/jme-07-0067.
- Kinyamu, H.K.; Collins, J.B.; Grissom, S.F.; Hebbar, P.B.; Archer, T.K. Genome wide transcriptional profiling in breast cancer cells reveals distinct changes in hormone receptor target genes and chromatin modifying enzymes after proteasome inhibition. Mol. Carcinog. 2008, 47, 845–885, doi:10.1002/mc.20440.
- Powers, G.L.; Ellison-Zelski, S.J.; Casa, A.J.; Lee, A.V.; Alarid, E.T. Proteasome inhibition represses ERα gene expression in ER+ cells: A new link between proteasome activity and estrogen signaling in breast cancer. Oncogene 2009, 29, 1509–1518, doi:10.1038/onc.2009.434.
- Powers, G.L.; Rajbhandari, P.; Solodin, N.M.; Bickford, B.; Alarid, E.T. The Proteasome Inhibitor Bortezomib Induces an Inhibitory Chromatin Environment at a Distal Enhancer of the Estrogen Receptor-α Gene. PLoS ONE 2013, 8, e81110, doi:10.1371/journal.pone.0081110.
- Berry, N.B.; Fan, M.; Nephew, K.P. Estrogen receptor-alpha hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol. Endocrinol. 2008, 22, 1535–1551, doi:10.1210/me.2007-0449.
- Tecalco-Cruz, A.C.; Ramirez-Jarquin, J.O.; Cruz-Ramos, E. Estrogen Receptor Alpha and its Ubiquitination in Breast Cancer Cells. Curr. Drug Targets 2019, 20, 690–704, doi:10.2174/1389450119666181015114041.