Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Amina Yu and Version 2 by Thien Vinh Luong.
The ketogen diet has been proposed as an effective intervention for type 2 diabetes and obesity since glycemic control is improved and sustained weight loss can be achieved. Interestingly, hyperketonemia is also associated with beneficial cardiovascular effects, possibly caused by improved cardiac energetics and reduced oxygen use. The objective of this narrative review is to provide insights into the ketogenic diet and its effects on myocardial ketone body utilization and, consequently, cardiovascular health.
- ketogenic diet
- ketone bodies
- heart
- metabolism
- heart failure
1. Ketone Bodies and the Heart
1.1. The Healthy Heart
The heart is often described as a metabolic omnivore since it readily utilizes all energy substrates as fuel and easily flexes between different compositions of energy substrates depending on energy demand and substrate availability (
1. Ketone Bodies and the Heart
1.1. The Healthy Heart
The heart is often described as a metabolic omnivore since it readily utilizes all energy substrates as fuel and easily flexes between different compositions of energy substrates depending on energy demand and substrate availability (
Figure 1). In the healthy, postabsorptive heart, fatty acids account for around 70% of cardiac energy consumption [19,20], whereas glucose and ketone bodies have been estimated to deliver less than 10% of the total energy demand [21]. Even this modest contribution from glucose and ketone bodies may be an overestimation, as evidenced by a recent elegant metabolomics’ study, in which glucose uptake was barely measurable and was estimated to account for less than 1% of total energy demand [22]. The remaining myocardial energy need is derived from oxidation of lactate and branched-chain amino acids [21,23]. Glucose, fatty acids, ketone bodies, and branched-chain amino acids are, thus, all sources of acetyl-CoA feeding into the Krebs cycle [14] ultimately generating ATP. Therefore, oxidation of a given substrate primarily occurs in proportion to the delivery of the substrate into the cardiomyocyte at the expense of other substrates [20]. In the postprandial state, carbohydrate consumption stimulates insulin secretion, resulting in increased myocardial glucose oxidation at the expense of fatty acid oxidation. This is mediated through (1) an increased glucose delivery through insulin-mediated glucose uptake in the cardiomyocyte, and (2) insulin-mediated inhibition of adipose tissue lipolysis with reduced fatty acid release and, therefore, also less fatty acid delivery to the heart. This was demonstrated in an elegant study by Ferrannini et al. [24], who used cardiac vein and arterial catherization during a hyperinsulinemic-euglycemic clamp to demonstrate that hyperinsulinemia dramatically increases myocardial uptake and utilization of glucose, lactate, and pyruvate. By contrast, myocardial extraction of circulating fatty acids, glycerol, and 3-OHB was almost completely absent. Interestingly, the shift from fat to carbohydrate metabolism did not change cardiac oxygen consumption, heart rate, blood flow, or cardiac pressure and work parameters. This flexibility ensures a constant supply of cardiac fuel even during rapidly changing hormonal and metabolic conditions. However, the study by Ferrannini et al. did not explore whether glucose is preferentially oxidized during conditions of ample supply of both carbohydrate and lipid fuel sources, including ketone bodies.
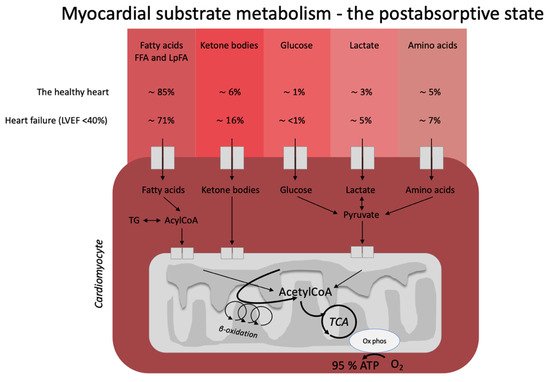
). In the healthy, postabsorptive heart, fatty acids account for around 70% of cardiac energy consumption [1][2], whereas glucose and ketone bodies have been estimated to deliver less than 10% of the total energy demand [3]. Even this modest contribution from glucose and ketone bodies may be an overestimation, as evidenced by a recent elegant metabolomics’ study, in which glucose uptake was barely measurable and was estimated to account for less than 1% of total energy demand [4]. The remaining myocardial energy need is derived from oxidation of lactate and branched-chain amino acids [3][5]. Glucose, fatty acids, ketone bodies, and branched-chain amino acids are, thus, all sources of acetyl-CoA feeding into the Krebs cycle [6] ultimately generating ATP. Therefore, oxidation of a given substrate primarily occurs in proportion to the delivery of the substrate into the cardiomyocyte at the expense of other substrates [2]. In the postprandial state, carbohydrate consumption stimulates insulin secretion, resulting in increased myocardial glucose oxidation at the expense of fatty acid oxidation. This is mediated through (1) an increased glucose delivery through insulin-mediated glucose uptake in the cardiomyocyte, and (2) insulin-mediated inhibition of adipose tissue lipolysis with reduced fatty acid release and, therefore, also less fatty acid delivery to the heart. This was demonstrated in an elegant study by Ferrannini et al. [7], who used cardiac vein and arterial catherization during a hyperinsulinemic-euglycemic clamp to demonstrate that hyperinsulinemia dramatically increases myocardial uptake and utilization of glucose, lactate, and pyruvate. By contrast, myocardial extraction of circulating fatty acids, glycerol, and 3-OHB was almost completely absent. Interestingly, the shift from fat to carbohydrate metabolism did not change cardiac oxygen consumption, heart rate, blood flow, or cardiac pressure and work parameters. This flexibility ensures a constant supply of cardiac fuel even during rapidly changing hormonal and metabolic conditions. However, Ferrannini et al. did not explore whether glucose is preferentially oxidized during conditions of ample supply of both carbohydrate and lipid fuel sources, including ketone bodies.
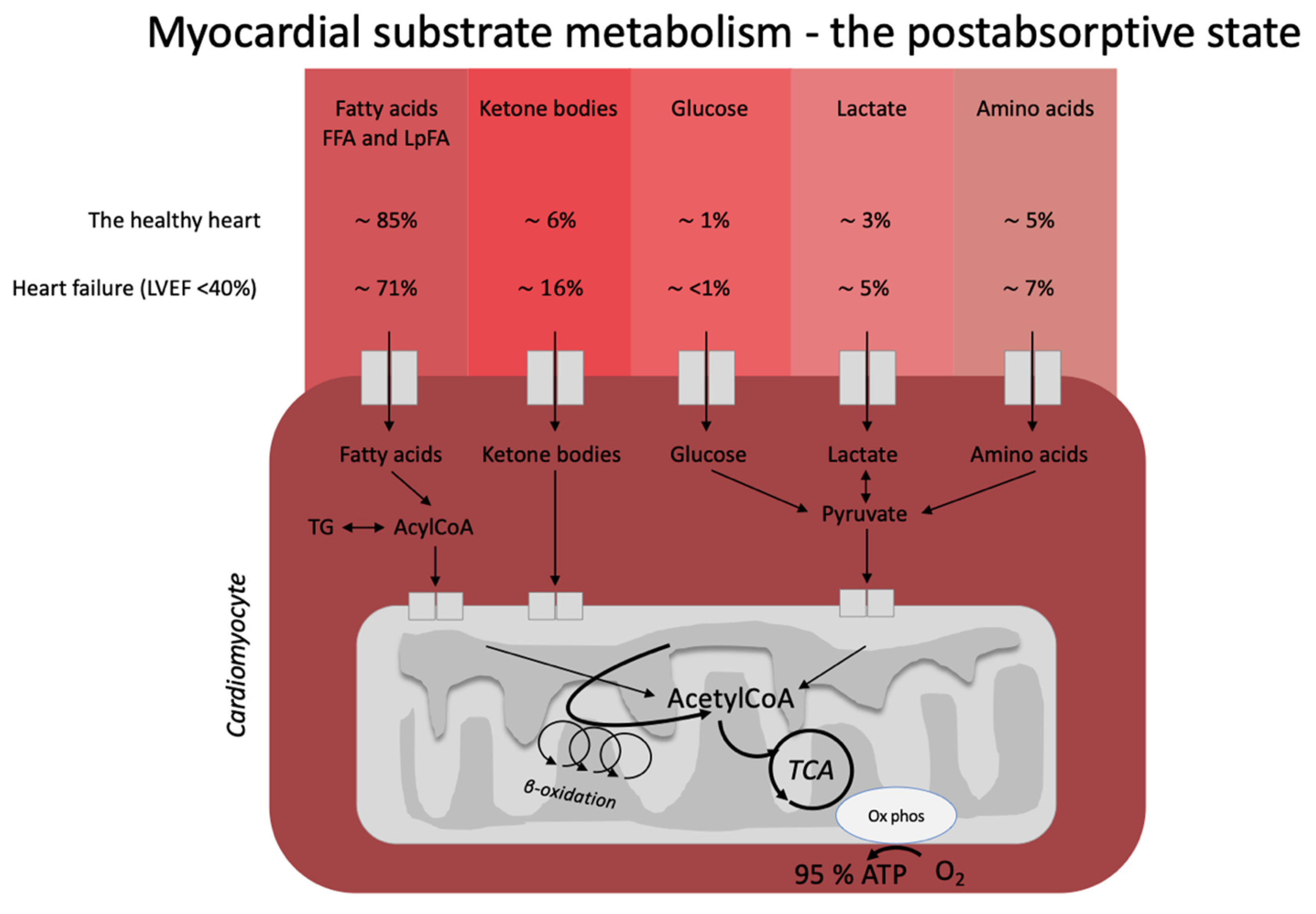
Figure 1. Postabsorptive myocardial substrate metabolism. In the healthy heart, the majority of energy expenditure arises from oxidation of fatty acids from FFA and circulating lipoproteins. In individuals with heart failure, oxidation of ketone bodies (3-hydroxybutyrate, acetoacetate) is upregulated at the expense of fatty acid oxidation. The estimates in the figure were extracted from the recent paper by Murashige et al. [22]. FFA: free fatty acids; LpFA: fatty acids from circulating lipoproteins; LVEF: left ventricle ejection fraction, ATP: adenosine triphosphate, TCA: tricarboxylic acid cycle, AcetylCoA: acetyl coenzyme A, TG: triglycerides.
1.2. Heart Failure
Heart failure is characterized by alterations in cardiac substrate metabolism, structural remodeling, and impaired contractility. The heart failure-related changes in myocardial metabolism are thought to be to some extent caused by mitochondrial oxidative dysfunction [23], resulting in a less effective utilization of energy substrates for ATP production. In addition, the failing heart becomes less flexible in shifting between energy substrates depending on their availability. The result is a sometimes paradoxically energy-starved [27] heart, fittingly described by wiser people as “an engine out of fuel” [28]. The failing heart is also characterized by impaired insulin signaling and insulin stimulation of glucose oxidation, resulting in a progressive development of cardiac insulin resistance [23]. As a consequence of the reduced ATP production by glucose oxidation, glycolytic processes are frequently upregulated, particularly evident in ischemic heart failure [29]. Moreover, fatty acid oxidation is decreased in tandem with the increase in glycolysis [21]. In this setting, it is becoming increasingly clear that the failing heart may rely much more on ketone bodies as a source of energy than the healthy heart [23,30]. In fact, it has been shown that ketone bodies account for 16% of total cardiac ATP generation in heart failure patients [22] and that the ketolytic enzymes BDH1 and SCOT are upregulated in the failing heart [31]. It is plausible that this is a metabolic adaptation to compensate for an otherwise ineffective substrate utilization and increased oxygen demand [32].
Ketone bodies may also have more direct effects on cardiac contractile function in individuals with heart failure. This was explored in a study by Nielsen et al., who demonstrated a dose-dependent increase in cardiac output of up to 2 L/min and an 8% increase in left ventricular ejection fraction as a result of an exogenous infusion of ketone bodies [11]. Somewhat disappointingly, however, the ketone body infusion did not improve myocardial external efficiency (MEE), which is impaired in heart failure and usually changes in a characteristic manner during medical or surgical cardiac interventions [33]. This lack of improvement in MEE implies that at least an acute elevation of ketone bodies may not have the anticipated oxygen-sparing effect. However, in that study MEE was measured during a hyperinsulinemic-euglycemic clamp and a sizable fraction of ATP production could, therefore, have been derived from glucose oxidation. It is possible that MEE could have been increased by an exogenous ketone body infusion performed in the postabsorptive state, where myocardial energy consumption is overwhelmingly served by relatively oxygen-inefficient fatty acids.
1.3. The Diabetic Heart Disease
The impact of ketone bodies on cardiac substrate metabolism has not been thoroughly studied in patients with diabetes. Diabetes is characterized by increased levels of circulating fatty acids and hyperglycemia; but, despite the increased delivery of glucose to the coronary circulation, the diabetic heart almost exclusively oxidizes fatty acids [34]. Much as in heart failure, the diabetic heart is metabolically inflexible and energetically inefficient, increasing the risk of myocardial injury during bouts of ischemia [14].
Individuals with diabetes may develop diabetic cardiomyopathy that does not necessarily resemble the typical cardiac pathology seen in patients with ischemic heart failure. Diabetic cardiomyopathy is characterized by early diastolic defects, interstitial fibrosis, and left ventricular hypertrophy followed by systolic dysfunction, which is then recognized as clinical heart failure. Diabetic cardiomyopathy can be observed in individuals with T2D even in the absence of the traditional risk factors for heart failure (hypertension, dyslipidemia, obesity, and coronary artery disease) [35,36] and carries a 2.5-fold increased risk of heart failure [37]. Independent risk factors for developing diabetic cardiomyopathy include insulin resistance, hyperinsulinemia, and hyperglycemia. It was hypothesized that diabetic cardiomyopathy is caused by inappropriate activation of the renin–angiotensin–aldosterone system, oxidative stress, lipotoxicity, inflammation, and dysfunctional immune modulation [38].
Overt diabetes is associated with markedly reduced insulin-mediated suppression of lipolysis (adipose tissue insulin resistance) [39], resulting in release of excess fatty acids that are converted to ketone bodies by the liver. Further adding to circulating ketone levels in diabetes, there are also indications that hepatic insulin resistance may result in increased hepatic ketogenesis [13]. Both individuals with and without type 2 diabetes take up ketone bodies in the heart at the expense of glucose, lactate, and pyruvate, as shown by Mizuno et al. using cardiac catherization of the coronary sinus and the aortic root [40]. It is conceivable that increased ketone body oxidation could serve as an oxygen-sparing adaption in the diabetic heart, which otherwise primarily oxidizes fatty acids. This shift in myocardial substrate oxidation is also one of the hypothesized benefits of SGLT-2 inhibitor treatment, where ketone levels increase to ~0.6 mM in individuals with type 2 diabetes [12]. Such a modest ketonemia does not closely mimic the levels of circulating ketone bodies achieved by infusions of ketone body salts (3–4 mM); it is, therefore, not surprising that 4 weeks of treatment with the SGLT2 inhibitor empagliflozin (resulting in ketone body levels of ~0.1–0.2 mM) does not affect PET-measured cardiac substrate metabolism or myocardial efficiency [41].
Postabsorptive myocardial substrate metabolism. In the healthy heart, the majority of energy expenditure arises from oxidation of fatty acids from FFA and circulating lipoproteins. In individuals with heart failure, oxidation of ketone bodies (3-hydroxybutyrate, acetoacetate) is upregulated at the expense of fatty acid oxidation. The estimates in the figure were extracted from the recent paper by Murashige et al. [4]. FFA: free fatty acids; LpFA: fatty acids from circulating lipoproteins; LVEF: left ventricle ejection fraction, ATP: adenosine triphosphate, TCA: tricarboxylic acid cycle, AcetylCoA: acetyl coenzyme A, TG: triglycerides.
1.2. Heart Failure
Heart failure is characterized by alterations in cardiac substrate metabolism, structural remodeling, and impaired contractility. The heart failure-related changes in myocardial metabolism are thought to be to some extent caused by mitochondrial oxidative dysfunction [5], resulting in a less effective utilization of energy substrates for adenosine triphosphate (ATP) production. In addition, the failing heart becomes less flexible in shifting between energy substrates depending on their availability. The result is a sometimes paradoxically energy-starved [8] heart, fittingly described by wiser people as “an engine out of fuel” [9]. The failing heart is also characterized by impaired insulin signaling and insulin stimulation of glucose oxidation, resulting in a progressive development of cardiac insulin resistance [5]. As a consequence of the reduced ATP production by glucose oxidation, glycolytic processes are frequently upregulated, particularly evident in ischemic heart failure [10]. Moreover, fatty acid oxidation is decreased in tandem with the increase in glycolysis [3]. In this setting, it is becoming increasingly clear that the failing heart may rely much more on ketone bodies as a source of energy than the healthy heart [5][11]. In fact, it has been shown that ketone bodies account for 16% of total cardiac ATP generation in heart failure patients [4] and that the ketolytic enzymes BDH1 and SCOT are upregulated in the failing heart [12]. It is plausible that this is a metabolic adaptation to compensate for an otherwise ineffective substrate utilization and increased oxygen demand [13].
Ketone bodies may also have more direct effects on cardiac contractile function in individuals with heart failure. This was explored in a study by Nielsen et al., who demonstrated a dose-dependent increase in cardiac output of up to 2 L/min and an 8% increase in left ventricular ejection fraction as a result of an exogenous infusion of ketone bodies [14]. Somewhat disappointingly, however, the ketone body infusion did not improve myocardial external efficiency (MEE), which is impaired in heart failure and usually changes in a characteristic manner during medical or surgical cardiac interventions [15]. This lack of improvement in MEE implies that at least an acute elevation of ketone bodies may not have the anticipated oxygen-sparing effect. However, in that study MEE was measured during a hyperinsulinemic-euglycemic clamp and a sizable fraction of ATP production could, therefore, have been derived from glucose oxidation. It is possible that MEE could have been increased by an exogenous ketone body infusion performed in the postabsorptive state, where myocardial energy consumption is overwhelmingly served by relatively oxygen-inefficient fatty acids.
1.3. The Diabetic Heart Disease
The impact of ketone bodies on cardiac substrate metabolism has not been thoroughly studied in patients with diabetes. Diabetes is characterized by increased levels of circulating fatty acids and hyperglycemia; but, despite the increased delivery of glucose to the coronary circulation, the diabetic heart almost exclusively oxidizes fatty acids [16]. Much as in heart failure, the diabetic heart is metabolically inflexible and energetically inefficient, increasing the risk of myocardial injury during bouts of ischemia [6].
Individuals with diabetes may develop diabetic cardiomyopathy that does not necessarily resemble the typical cardiac pathology seen in patients with ischemic heart failure. Diabetic cardiomyopathy is characterized by early diastolic defects, interstitial fibrosis, and left ventricular hypertrophy followed by systolic dysfunction, which is then recognized as clinical heart failure. Diabetic cardiomyopathy can be observed in individuals with T2D even in the absence of the traditional risk factors for heart failure (hypertension, dyslipidemia, obesity, and coronary artery disease) [17][18] and carries a 2.5-fold increased risk of heart failure [19]. Independent risk factors for developing diabetic cardiomyopathy include insulin resistance, hyperinsulinemia, and hyperglycemia. It was hypothesized that diabetic cardiomyopathy is caused by inappropriate activation of the renin–angiotensin–aldosterone system, oxidative stress, lipotoxicity, inflammation, and dysfunctional immune modulation [20].
Overt diabetes is associated with markedly reduced insulin-mediated suppression of lipolysis (adipose tissue insulin resistance) [21], resulting in release of excess fatty acids that are converted to ketone bodies by the liver. Further adding to circulating ketone levels in diabetes, there are also indications that hepatic insulin resistance may result in increased hepatic ketogenesis [22]. Both individuals with and without type 2 diabetes take up ketone bodies in the heart at the expense of glucose, lactate, and pyruvate, as shown by Mizuno et al. using cardiac catherization of the coronary sinus and the aortic root [23]. It is conceivable that increased ketone body oxidation could serve as an oxygen-sparing adaption in the diabetic heart, which otherwise primarily oxidizes fatty acids. This shift in myocardial substrate oxidation is also one of the hypothesized benefits of SGLT-2 inhibitor treatment, where ketone levels increase to ~0.6 mM in individuals with type 2 diabetes [24]. Such a modest ketonemia does not closely mimic the levels of circulating ketone bodies achieved by infusions of ketone body salts (3–4 mM); it is, therefore, not surprising that 4 weeks of treatment with the SGLT2 inhibitor empagliflozin (resulting in ketone body levels of ~0.1–0.2 mM) does not affect PET-measured cardiac substrate metabolism or myocardial efficiency [25].
2. The Ketogenic Diet and the Heart
2. The Ketogenic Diet and the Heart
Whereas pharmacological intervention may increase circulating ketone bodies to a modest degree, dietary interventions may potentially result in vastly more pronounced and sustained hyperketonemia. Of these, the KD has been investigated most frequently. The KD entails a strict reduction in carbohydrate intake to reduce insulin secretion and, by extension, insulin-mediated inhibition of ketogenesis. The diet belongs to a group of low-carbohydrate diets (LCD), which usually provide 50–150 g of carbohydrates, equivalent to 10–30% of the daily caloric intake. Diets with higher amounts of carbohydrate intake do not impact ketone body levels and will not be discussed further [42]. One of the main problems evaluating the effect of LCD has turned out to be a lack of diet standardization. Numerous diets are within the definitions of LCD but with substantial differences in fat and protein content as well as total caloric content. The classic KD contains much fat (55 to 90% of total calories), moderate amounts of protein (15–35% of total calories), and a very low carbohydrate content (5–10% of total calories) [43,44,45]. Whereas most dietary fats from standard foods are long-chain triglycerides (LCT), medium-chained triglycerides (MCT) primarily containing octanoic and decanoic acid yield more ketone bodies per kilocalorie of energy compared to LCT [46]. In addition, fatty acids from MCT are absorbed directly and delivered to the liver through the portal system. However, MCT’s can only be ingested as supplements in the form of oils and can, therefore, not be labeled a diet in itself. In addition, the ketogenic potential of ingesting MCT is often attenuated by the accompanying ingestion of carbohydrates and protein. In the modified Atkins diet (MAD), around 50–65% of the calories are derived from standard fat sources [47] with a larger protein intake compared to the KD. The MAD is more palatable and less restrictive than the KD, thus increasing compliance in patients with behavioral problems and children treated by diets for epilepsy [46]. However, the greater protein content of the MAD reduces the ketogenic effect, since amino acids from the ingested protein stimulate insulin secretion and thereby inhibit ketogenesis [48]. Furthermore, the ingested amino acids are also converted into glucose through gluconeogenesis, which can lead to higher glucose levels and stimulation of insulin secretion. Since no clear consensus exists regarding the specific quantity and quality of each macronutrient in LCD, it is difficult to compare the different studies and determine to what extent these different diets impact substrate metabolism. Overall, the KD is the most carbohydrate restrictive diet, providing less than 50 g and sometimes even less than 20 g of carbohydrates per day. This results in a greater increase in ketone bodies than what is observed during other LCDs [42,43]. In fact, 3-beta-hydroxy-butyrate (3-OHB) may increase to 3.2 mM after only 4 days on a KD [49] and to 5.2 mM for children on a 12-month classical, strict KD [50].
Whereas pharmacological intervention may increase circulating ketone bodies to a modest degree, dietary interventions may potentially result in vastly more pronounced and sustained hyperketonemia. Of these, the KD has been investigated most frequently. The ketogenic diet (KD) entails a strict reduction in carbohydrate intake to reduce insulin secretion and, by extension, insulin-mediated inhibition of ketogenesis. The diet belongs to a group of low-carbohydrate diets (LCD), which usually provide 50–150 g of carbohydrates, equivalent to 10–30% of the daily caloric intake. Diets with higher amounts of carbohydrate intake do not impact ketone body levels and will not be discussed further [26]. One of the main problems evaluating the effect of LCD has turned out to be a lack of diet standardization. Numerous diets are within the definitions of LCD but with substantial differences in fat and protein content as well as total caloric content. The classic KD contains much fat (55 to 90% of total calories), moderate amounts of protein (15–35% of total calories), and a very low carbohydrate content (5–10% of total calories) [27][28][29]. Whereas most dietary fats from standard foods are long-chain triglycerides (LCT), medium-chained triglycerides (MCT) primarily containing octanoic and decanoic acid yield more ketone bodies per kilocalorie of energy compared to LCT [30]. In addition, fatty acids from MCT are absorbed directly and delivered to the liver through the portal system. However, MCT’s can only be ingested as supplements in the form of oils and can, therefore, not be labeled a diet in itself. In addition, the ketogenic potential of ingesting MCT is often attenuated by the accompanying ingestion of carbohydrates and protein. In the modified Atkins diet (MAD), around 50–65% of the calories are derived from standard fat sources [31] with a larger protein intake compared to the KD. The MAD is more palatable and less restrictive than the KD, thus increasing compliance in patients with behavioral problems and children treated by diets for epilepsy [30]. However, the greater protein content of the MAD reduces the ketogenic effect, since amino acids from the ingested protein stimulate insulin secretion and thereby inhibit ketogenesis [32]. Furthermore, the ingested amino acids are also converted into glucose through gluconeogenesis, which can lead to higher glucose levels and stimulation of insulin secretion. Since no clear consensus exists regarding the specific quantity and quality of each macronutrient in LCD, it is difficult to compare the different studies and determine to what extent these different diets impact substrate metabolism. Overall, the KD is the most carbohydrate restrictive diet, providing less than 50 g and sometimes even less than 20 g of carbohydrates per day. This results in a greater increase in ketone bodies than what is observed during other LCDs [26][27]. In fact, 3-beta-hydroxy-butyrate (3-OHB) may increase to 3.2 mM after only 4 days on a KD [33] and to 5.2 mM for children on a 12-month classical, strict KD [34].