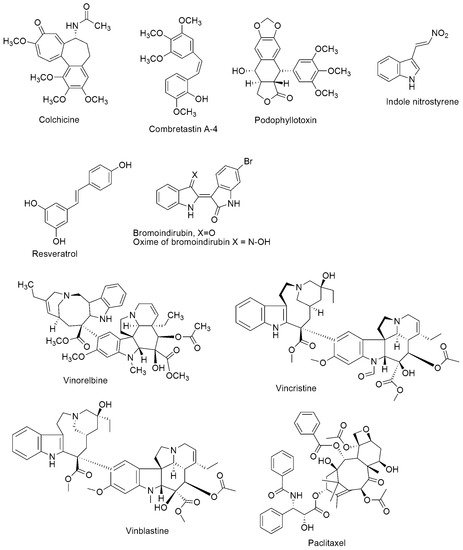
Microtubules are cylindrical protein polymers formed from αβ-tubulin heterodimers in the cytoplasm of eukaryotic cells. Microtubule disturbance may cause cell cycle arrest in the G2/M phase, and anomalous mitotic spindles will form. Microtubules are an important target for cancer drug action because of their critical role in mitosis. Several microtubule-targeting agents with vast therapeutic advantages have been developed, but they often lead to multidrug resistance and adverse side effects. Thus, single-target therapy has drawbacks in the effective control of tubulin polymerization. Molecular hybridization, based on the amalgamation of two or more pharmacophores of bioactive conjugates to engender a single molecular structure with enhanced pharmacokinetics and biological activity, compared to their parent molecules, has recently become a promising approach in drug development. The practical application of combined active scaffolds targeting tubulin polymerization inhibitors has been corroborated in the past few years
1. Introduction
Microtubules are cylindrical protein polymers assembled in the cytoplasm of all eukaryotic cells by polymerization of αβ-tubulin heterodimers. Microtubules are characterized by a unique dynamic instability process, which involves continuously alternating phases of elongation and shortening interspersed with catastrophic disassembly events. In living cells, the minus ends of microtubules are associated with structures called microtubule organizing centers (MTOCs) [1][3]. The main MTOC in the cell is the centrosome, contiguously positioned to the nucleus. Microtubules play an essential function in the maintenance of cell shape and in the progression of cell division [2][4]. They are the principal components for the formation of the mitotic spindles. They all participate in signaling, intracellular transport, and cell motility [3][5]. Disturbance of microtubules may induce cell cycle arrest at the G2/M phase. Their pre-eminent importance in force generation in mitosis to enable the segregation of chromosomes makes microtubules attractive as intra-cellular targets for anti-cancer drug action.
Drugs that impede the mitotic spindle apparatus have been the mainstay of effective cancer chemotherapy. They are referred to as tubulin-binding agents, antimicrotubular, or anti-tubulin compounds (see
Figure 1). They have drawn much interest in the past two decades, making microtubules important drug targets. Several natural compounds such as paclitaxel, epothilones, vinblastine, combretastatin, and colchicine act by altering microtubule dynamics. Besides, compounds originating from different medicinal chemistry strategies can be classified based on their mechanism of action and the location of the primary binding site. Tubulin polymerization inhibitors can interact with residues in either the colchicine binding site or Vinca alkaloids binding site, while the tubulin depolymerization inhibitors interact with the taxane site. These drugs have significantly improved survivorship rates and management of several cancer types among children and adults. Nevertheless, the intrinsic or acquired multidrug resistance (MDR) [4][5][6], as well as numerous adverse side effects such as chemotherapy-induced peripheral neuropathy, nausea, vomiting, diarrhea, constipation, paralytic ileus, urinary retention, bone marrow suppression, and neurotoxicity [7][8][9] of these drugs have been of significant concern. As a result, toxicity and drug resistance are the main properties requiring constant improvement when developing novel tubulin polymerization inhibitors with acceptable pharmacological profiles. Additionally, combination therapy has been a strategy aimed at enhancing the potency of several drugs such that one drug can impede the growth of cancer cells in a specific phase, and another cancer agent can function in a different phase. In addition to the complex regimens that use multiple medications, the combination of cancer chemotherapy with surgery reduces the number of cancer cells and radiotherapy destroys cancer cells even further. Hence, many cytotoxic hybrids have been clustered into regimens of anticancer behavior that improve treatment outcomes with fewer side effects. However, combination therapy is often non-advantageous due to poor patient adherence as well as the risk of overlapping toxicities. 2). They have drawn much interest in the past two decades, making microtubules important drug targets. Several natural compounds such as paclitaxel, epothilones, vinblastine, combretastatin, and colchicine act by altering microtubule dynamics. Besides, compounds originating from different medicinal chemistry strategies can be classified based on their mechanism of action and the location of the primary binding site. Tubulin polymerization inhibitors can interact with residues in either the colchicine binding site or Vinca alkaloids binding site, while the tubulin depolymerization inhibitors interact with the taxane site. These drugs have significantly improved survivorship rates and management of several cancer types among children and adults. Nevertheless, the intrinsic or acquired multidrug resistance (MDR) [6,7,8], as well as numerous adverse side effects such as chemotherapy-induced peripheral neuropathy, nausea, vomiting, diarrhea, constipation, paralytic ileus, urinary retention, bone marrow suppression, and neurotoxicity [9,10,11] of these drugs have been of significant concern. As a result, toxicity and drug resistance are the main properties requiring constant improvement when developing novel tubulin polymerization inhibitors with acceptable pharmacological profiles. Additionally, combination therapy has been a strategy aimed at enhancing the potency of several drugs such that one drug can impede the growth of cancer cells in a specific phase, and another cancer agent can function in a different phase. In addition to the complex regimens that use multiple medications, the combination of cancer chemotherapy with surgery reduces the number of cancer cells and radiotherapy destroys cancer cells even further. Hence, many cytotoxic hybrids have been clustered into regimens of anticancer behavior that improve treatment outcomes with fewer side effects. However, combination therapy is often non-advantageous due to poor patient adherence as well as the risk of overlapping toxicities.
Figure 12. Chemical structures of tubulin polymerization inhibitors.
2. Tubulin Polymerization Hybrids
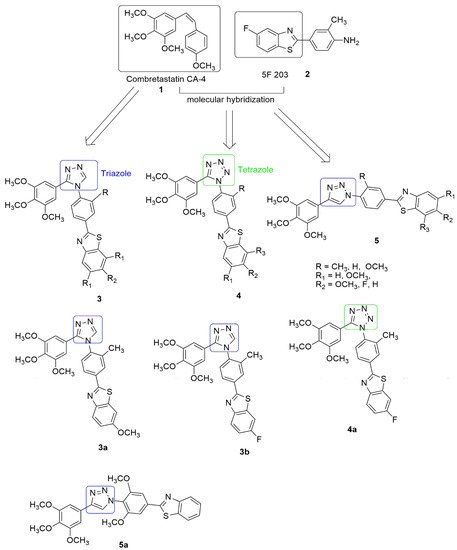
Triazole and Tetrazole Hybrids
Triazole occurs as an isomer depending on the position of the three nitrogen atoms in the ring. The two possible isomers include 1,2,3-triazole and 1,2,4-triazole. Triazole is highly versatile with a broad spectrum of biological activities. It has been introduced in several clinical drugs [10][11], including cancer drugs [12][13][14] (letrozole and anastrozole), thus drawing attention to the importance of this nucleus. Tetrazole is a useful building block commonly used in developing new drugs due to its metabolic stability, the non-metabolized bio-isosteric analog of carboxylic, cis-amide, and other expedient physicochemical properties. Furthermore, the isomer forms of tetrazole such as 1H and 2H-tetrazole have been reported to be effective for designing anti-cancer compounds. Combretastatin (CA-4) Chemical structures of tubulin polymerization inhibitors.
2. Tubulin Polymerization Hybrids
Triazole and Tetrazole Hybrids
Triazole occurs as an isomer depending on the position of the three nitrogen atoms in the ring. The two possible isomers include 1,2,3-triazole and 1,2,4-triazole. Triazole is highly versatile with a broad spectrum of biological activities. It has been introduced in several clinical drugs [12,13], including cancer drugs [14,15,16] (letrozole and anastrozole), thus drawing attention to the importance of this nucleus. Tetrazole is a useful building block commonly used in developing new drugs due to its metabolic stability, the non-metabolized bio-isosteric analog of carboxylic, cis-amide, and other expedient physicochemical properties. Furthermore, the isomer forms of tetrazole such as 1H and 2H-tetrazole have been reported to be effective for designing anti-cancer compounds. Combretastatin (CA-4) has effectively inhibited tumor cell growth, such as MDR cancer cell lines, and displays significant inhibition of tubulin polymerization and excellent cytotoxicity against murine lymphocytic leukemia human ovarian and colon cancer cell lines. However, it lacks water solubility. On the other hand, 5F-203,
2 and its prodrug, phortress, are aqueous soluble and chemically stable, while the clinical evaluation of phortress displayed selective antitumor activity. Interestingly, Rao et al. [15]. synthesized cis restricted triazole/tetrazole analogue of CA-4 and benzothiazole scaffolds and its prodrug, phortress, are aqueous soluble and chemically stable, while the clinical evaluation of phortress displayed selective antitumor activity. Interestingly, Rao et al. [17]. synthesized cis restricted triazole/tetrazole analogue of CA-4 and benzothiazole scaffolds . The cis configuration of the olefinic bond was restricted by incorporating triazole and tetrazole rings. The synthesized compounds were tested against selected human cancer cell lines, namely, prostate (DU-145), cervix (HeLa), lung adenocarcinoma (A549), liver (HepG2), and breast (MCF-7), using CA-4 as the reference compound. In the compounds with 1,2,4-triazole (
with a methyl substitution on the C′-3 position of the 2-phenylbenzothiazole moiety and OCH3 and F, substitution on the C′-6 position of benzothiazole moiety displayed great inhibitory activity with an IC50 value of 0.054 and 0.048 µM against the lung cancer cell line. These compounds also cause the arrest of cells in the G2/M phase and induce apoptosis in A549 cells by activation of caspase 3. They significantly inhibit tubulin assembly with IC50 values of 1.67 and 1.00 µM and bind to the colchicine site of beta (β) tubulin. The substitution of 1,2,4-triazole linker with 1,2,3,4- tetrazole (
) resulted in moderate activity.
Figure 2 displays the most potent compound in the 1,2,3,4-tetrazole (
3 displays the most potent compound in the 1,2,3,4-tetrazole (
) group with IC50 values of 0.246 and 0.243 µM, respectively, against the lung cancer cell line. In summary, derivatives bearing phenyl ring attached to benzothiazole ring (R = Me) at C′3-position significantly increased the activity compared to other substituents. Based on the molecular docking, the pose view of
fits well in the colchicine binding domain. The tri-OCH3 phenyl group has been found deep in the hydrophobic site of the β chain, and it is in close contact with amino acids, Val238, Thr239, Cys241, Leu242, Ala250, Leu252, Leu255, Ala316, Ala317, Val318, Thr353, Ala354, and Ile378, respectively. The benzothiazole is extended towards the α, β interface of the tubulin and forms close contact with Gln11, Ser178, and Tyr224 of the α chain and Glu247 and Gln248 residues of the β chain. However, 1-phenytriazole is in close contact with Asn101, Ala180, and Val181 residues of a chain and Asn258, Met259, and Lys352 residues of the β chain.
Figure 23. Chemical structures of triazole/tetrazole–benzothiazole hybrids with anti-tubulin activities.
Xu et al. [16] synthesized series of 3,6-diaryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines by replacing the (Z,E)-butadiene the spacer of vinylogous CA-4 Chemical structures of triazole/tetrazole–benzothiazole hybrids with anti-tubulin activities.
Xu et al. [18] synthesized series of 3,6-diaryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines by replacing the (Z,E)-butadiene the spacer of vinylogous CA-4 with a novel rigid [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine moiety. Subsequently, the antiproliferative activity of the compounds was tested against three human cancer cell lines, including SGC-7901, A549 (gastric adenocarcinoma), and HT-1080 (fibrosarcoma) cells, using MTT assay and CA-4 was utilized as the positive control. Out of the 32 synthesized compounds tested for their antiproliferative activities. Compound
7a showed the most powerful antiproliferative activities against SGC-7901, A549, and HT-1080 cell lines with an IC50 of 0.011–0.015 μM, compared to the standard drug CA-4 (0.009–0.013 μM).
Piperazine has been reported as a potent compound that obstructs mitosis, induces cell apoptosis, binds to the colchicine site, and inhibits the tubulin [17][18]. Meanwhile, sulfonamide compounds are antibacterial, anticancer, antithyroid, antidiabetic, antiviral, and antihypertensive agents.
Sulfanilamide skeleton is a bioactive unit that exhibits strong anti-tumor activity [19][20]. Due to the anti-tumor activity of sulfanilamide and triazole, Guo et al. [21] projected that a sulfanilamide showed the most powerful antiproliferative activities against SGC-7901, A549, and HT-1080 cell lines with an IC50 of 0.011–0.015 μM, compared to the standard drug CA-4 (0.009–0.013 μM).
Piperazine has been reported as a potent compound that obstructs mitosis, induces cell apoptosis, binds to the colchicine site, and inhibits the tubulin [19,20]. Meanwhile, sulfonamide compounds are antibacterial, anticancer, antithyroid, antidiabetic, antiviral, and antihypertensive agents.
Sulfanilamide skeleton is a bioactive unit that exhibits strong anti-tumor activity [26,27]. Due to the anti-tumor activity of sulfanilamide and triazole, Guo et al. [28] projected that a sulfanilamide ring linked to a 1,2,3-triazole
unit would serve as novel and effective tubulin polymerization inhibitors. The synthesis and antiproliferative activity of sulfanilamide-1,2,3-triazole hybrids
against three selected human cancer cell lines, namely, BGC-823 (gastric), MGC-803 (gastric carcinoma), and SGC-7901, were reported. Compound
showed the most excellent inhibitory effect against MGC-803 cells, with an IC50 value of 0.4 ± 0.1 μM.
Podophyllotoxin (PPT)
17 is a non-alkaloid toxin lignin isolated from Podophyllum peltatum L. and Podophyllum hexandrum, and it possesses a large variety of medical applications. Etoposide and teniposide are semi-synthetic derivatives of podophyllotoxin and differ significantly in their mode of action. These compounds are DNA topoisomerase II inhibitors, and the inhibition of topoisomerase II blocks DNA from separating. Podophyllotoxin can successfully impede the microtubules construction. Their shortcomings include a lack of water solubility and drug resistance. The modifications of C-4 position have shown excellent inhibitory activity. In this regard, a series of triazolo-4β-amidopodophyllotoxin derivatives using an amide spacer were synthesized and examined for their cytotoxic activity against four human cancer cell lines, namely MCF-7 (breast cancer), B16 (oral cancer), HT 29 (colon cancer), and HeLa (cervical cancer) as reported by Vishnuvardhan et al. [22].
A combination of the structure of benzo [b]furan,
is a non-alkaloid toxin lignin isolated from Podophyllum peltatum L. and Podophyllum hexandrum, and it possesses a large variety of medical applications. Etoposide and teniposide are semi-synthetic derivatives of podophyllotoxin and differ significantly in their mode of action. These compounds are DNA topoisomerase II inhibitors, and the inhibition of topoisomerase II blocks DNA from separating. Podophyllotoxin can successfully impede the microtubules construction. Their shortcomings include a lack of water solubility and drug resistance. The modifications of C-4 position have shown excellent inhibitory activity. In this regard, a series of triazolo-4β-amidopodophyllotoxin derivatives using an amide spacer were synthesized and examined for their cytotoxic activity against four human cancer cell lines, namely MCF-7 (breast cancer), B16 (oral cancer), HT 29 (colon cancer), and HeLa (cervical cancer) as reported by Vishnuvardhan et al. [29].
A combination of the structure of benzo [b]furan,
to produce a single molecule has been explored. The hybrid compounds containing benzo[b]furan and triazole moieties (
22) with their antiproliferative activities against four-panel of human cancer cell lines (HCT116 colon cancer, HepG2 hepatic carcinoma, HeLa human epithelial cervical cancer, and A549 non-small-cell lung cancer) were reported by Qi et al. [23]. The 6-methoxy bearing ring’s antiproliferative activities were superior to the unsubstituted compounds. Compound ) with their antiproliferative activities against four-panel of human cancer cell lines (HCT116 colon cancer, HepG2 hepatic carcinoma, HeLa human epithelial cervical cancer, and A549 non-small-cell lung cancer) were reported by Qi et al. [30]. The 6-methoxy bearing ring’s antiproliferative activities were superior to the unsubstituted compounds. Compound exhibited the most excellent antiproliferative activities against all the tested cell lines, with IC50 values of 0.87 ± 0.79 (HCT116), 0.73 ± 0.67 (HeLa), 5.74 ± 1.21 (HepG2), and 0.57 ± 0.31 µM, respectively.
A novel series of tubulin inhibitors bearing an indole-1,2,4-triazole (
25) scaffold have been synthesized, and their biological activity has been reported [24]. Among the 18 compounds evaluated for anti-proliferation activity against HepG2 (human hepatoma cells), HeLa (human cervix cell), MCF-7 (breast cancer cell), and A549 (human lung cancer cell), compound ) scaffold have been synthesized, and their biological activity has been reported [31]. Among the 18 compounds evaluated for anti-proliferation activity against HepG2 (human hepatoma cells), HeLa (human cervix cell), MCF-7 (breast cancer cell), and A549 (human lung cancer cell), compound displayed excellent activity with IC50 values of 0.23 ± 0.08, 0.15 ± 0.18, 0.38 ± 0.12, and 0.30 ± 0.13 μM, respectively. The substitution of electron-donating groups such as OCH3 and CH3 (
) enhanced the anti-proliferative activity compared to the electron-withdrawing groups (F, Cl, Br, I, and CF3). The results from the tubulin polymerization assay showed that compound
was the most potent anti-tubulin agent (IC50 = 2.1 ± 0.12 μM), superior to the inhibitory activity of colchicine (2.52 ± 0.23 μM).
The pharmacophoric imidazopyridine
29 moiety have been found to have a wide range of biological activities. This scaffold has been explored to design new hybrids of antimicrobial, anticonvulsant, antipyretics, anti-inflammatory, anticancer, and many others. Imidazopyridine-guanylhydrazone displayed strong anticancer activity against different cancer cell lines. The derivative also arrested cell cycle in G2/M phase on SK-LU-1 cell line, downregulate cyclin-D1, E1, CDK2, and caused activation of Caspase-3 [25][26]. Because of this, Sayeed and co-workers combined triazole moiety have been found to have a wide range of biological activities. This scaffold has been explored to design new hybrids of antimicrobial, anticonvulsant, antipyretics, anti-inflammatory, anticancer, and many others. Imidazopyridine-guanylhydrazone displayed strong anticancer activity against different cancer cell lines. The derivative also arrested cell cycle in G2/M phase on SK-LU-1 cell line, downregulate cyclin-D1, E1, CDK2, and caused activation of Caspase-3 [35,36]. Because of this, Sayeed and co-workers combined triazole 29 group into a single entity [27]. They were subsequently evaluated for their cytotoxic effect in human cancer cells, precisely, lung (A549), prostate (DU-145), colon (HCT-116), and breast (MDA-MB 231). Two conjugates, group into a single entity [37]. They were subsequently evaluated for their cytotoxic effect in human cancer cells, precisely, lung (A549), prostate (DU-145), colon (HCT-116), and breast (MDA-MB 231). Two conjugates, 31b (IC50 = 0.51 and 0.63 μM) were found to be more active than the remaining conjugates against A549 [27]. These conjugates caused cell cycle arrest at the G2/M phase, signifying inhibition of tubulin polymerization and induced apoptosis through the mitochondrial pathway.
3. Benzimidazole Hybrids
Benzimidazole is a privileged heterocyclic scaffold found in many natural and pharmacologically functional molecules. Benzimidazole, also known as 3-azaindole, azindole, benziminazole, benzoglyoxaline, 3-benzodiazole, and 1,3-diazaindene; containing fusion of benzene and imidazole. The organic molecule is colorless, freely soluble in alcohol, melts at 170.5 °C, and boils at 360 °C. The core structure of benzimidazole is a vital pharmacophore at present. It has been used as a preferred scaffold for synthesizing selected drugs of interest in medicinal chemistry, including antimicrobial [28][29], analgesic [30], antihelmintic [31][32], antioxidant [33], antimalarial [34], antiviral [35], and anticancer [36] activity. Many benzimidazole-based derivatives are available as medicines such as bendamustine, veliparib, nocodazole, liarozole, and pracinosta. Further, dehydroabietic acid is a natural diterpenic resin acid, and its derivatives exhibited strong anticancer activity through different mechanisms of action (IC50 = 0.51 and 0.63 μM) were found to be more active than the remaining conjugates against A549 [37]. These conjugates caused cell cycle arrest at the G2/M phase, signifying inhibition of tubulin polymerization and induced apoptosis through the mitochondrial pathway.
3. Benzimidazole Hybrids
Benzimidazole is a privileged heterocyclic scaffold found in many natural and pharmacologically functional molecules. Benzimidazole, also known as 3-azaindole, azindole, benziminazole, benzoglyoxaline, 3-benzodiazole, and 1,3-diazaindene; containing fusion of benzene and imidazole. The organic molecule is colorless, freely soluble in alcohol, melts at 170.5 °C, and boils at 360 °C. The core structure of benzimidazole is a vital pharmacophore at present. It has been used as a preferred scaffold for synthesizing selected drugs of interest in medicinal chemistry, including antimicrobial [39,40], analgesic [41], antihelmintic [42,43], antioxidant [44], antimalarial [45], antiviral [46], and anticancer [47] activity. Many benzimidazole-based derivatives are available as medicines such as bendamustine, veliparib, nocodazole, liarozole, and pracinosta. Further, dehydroabietic acid is a natural diterpenic resin acid, and its derivatives exhibited strong anticancer activity through different mechanisms of action . The 3,4,5-trimethoxyphenyl (TMP)
37 sub-unit were a remarkable pharmacophoric group of tubulin inhibitors. The units can be combined to generate a new series of dual-acting action hybrids.
ABT-751 [37] sub-unit were a remarkable pharmacophoric group of tubulin inhibitors. The units can be combined to generate a new series of dual-acting action hybrids.
are sulfonamide-bearing compounds with strong anti-tubulin and antimitotic properties. Due to the interesting properties of sulfonamide
44 and benzimidazole, Wang et al. [39]. focused on synthesizing a new series of 1-phenylsulphonyl-2-(1-methylindol-3-yl)-benzimidazole derivatives ( and benzimidazole, Wang et al. [50]. focused on synthesizing a new series of 1-phenylsulphonyl-2-(1-methylindol-3-yl)-benzimidazole derivatives ( 45). The compounds were further probed for anti-cancer against human cancer cell lines, specifically, A549 (lung), Hela (cervical), HepG2 (liver), and MCF-7 (breast).
The anti-mutagenicity effect of cinnamic acid lies in its ability to network with cellular nucleophiles such as glutathione (GSH) and cysteine. At the same time, the existence of a,ß-unsaturated carbonylic functional unit is liable for cellular interactions of cinnamic acid [40]. Donthiboina et al. [40] reported a synthetic route to benzimidazole and cinnamic acid hybrids ). The compounds were further probed for anti-cancer against human cancer cell lines, specifically, A549 (lung), Hela (cervical), HepG2 (liver), and MCF-7 (breast).
The anti-mutagenicity effect of cinnamic acid lies in its ability to network with cellular nucleophiles such as glutathione (GSH) and cysteine. At the same time, the existence of a,ß-unsaturated carbonylic functional unit is liable for cellular interactions of cinnamic acid [52]. Donthiboina et al. [52] reported a synthetic route to benzimidazole and cinnamic acid hybrids using a molecular hybridization approach, and the synthesized compounds were screened for their cytotoxic activity. Among the tested compounds,
displayed potent cytotoxic activity against lung cancer cell line with IC50 value of 0.29 ± 0.02 μM and showed less cytotoxicity when tested on NRK-52E (normal rat kidney epithelial cell line) with IC50 value of 1.58 ± 0.43 μM. The SAR showed a lack of substitution on the phenyl ring of cinnamides (R1 = H), resulting in a reduction in cytotoxicity. The insertion of electron-withdrawing groups (R or R1 = F, Cl, Br, and NO2), on the phenyl ring of benzimidazole or cinnamide led to either reduced or lost activity. In contrast, electron-rich substitution on either of the rings resulted in moderate to good activity. Besides, substituting 3,4,5-tri OCH3 on the phenyl ring of cinnamide (
, IC50 values >30 μM) reduced the activity due to steric hindrance. Compound
51a induced apoptosis by producing ROS, inhibited tubulin polymerization (IC50 value = 4.64 ± 0.09 μM), and changed the cell cycle by blocking progression at the G2/M phase.
A series of thioether and amine-bridged cinnamide derived pyrimidine-benzimidazole hybrids were designed based on the pharmacophore hybridization approach by mimicking the structural similarities of reported anti-tubulin agents [41]. The SAR analysis showed that substitution featuring bulkier groups such as 2-bromo-4,6-dimethoxy and 2-bromo-4- methoxy on the cinnamide scaffold were stable in both the molecular series ( induced apoptosis by producing ROS, inhibited tubulin polymerization (IC50 value = 4.64 ± 0.09 μM), and changed the cell cycle by blocking progression at the G2/M phase.
A series of thioether and amine-bridged cinnamide derived pyrimidine-benzimidazole hybrids were designed based on the pharmacophore hybridization approach by mimicking the structural similarities of reported anti-tubulin agents [53]. The SAR analysis showed that substitution featuring bulkier groups such as 2-bromo-4,6-dimethoxy and 2-bromo-4- methoxy on the cinnamide scaffold were stable in both the molecular series ( ). Among the 31 compounds tested for anti-cancer activity,
59b with CF3 group at position two of benzimidazole has the highest potency.
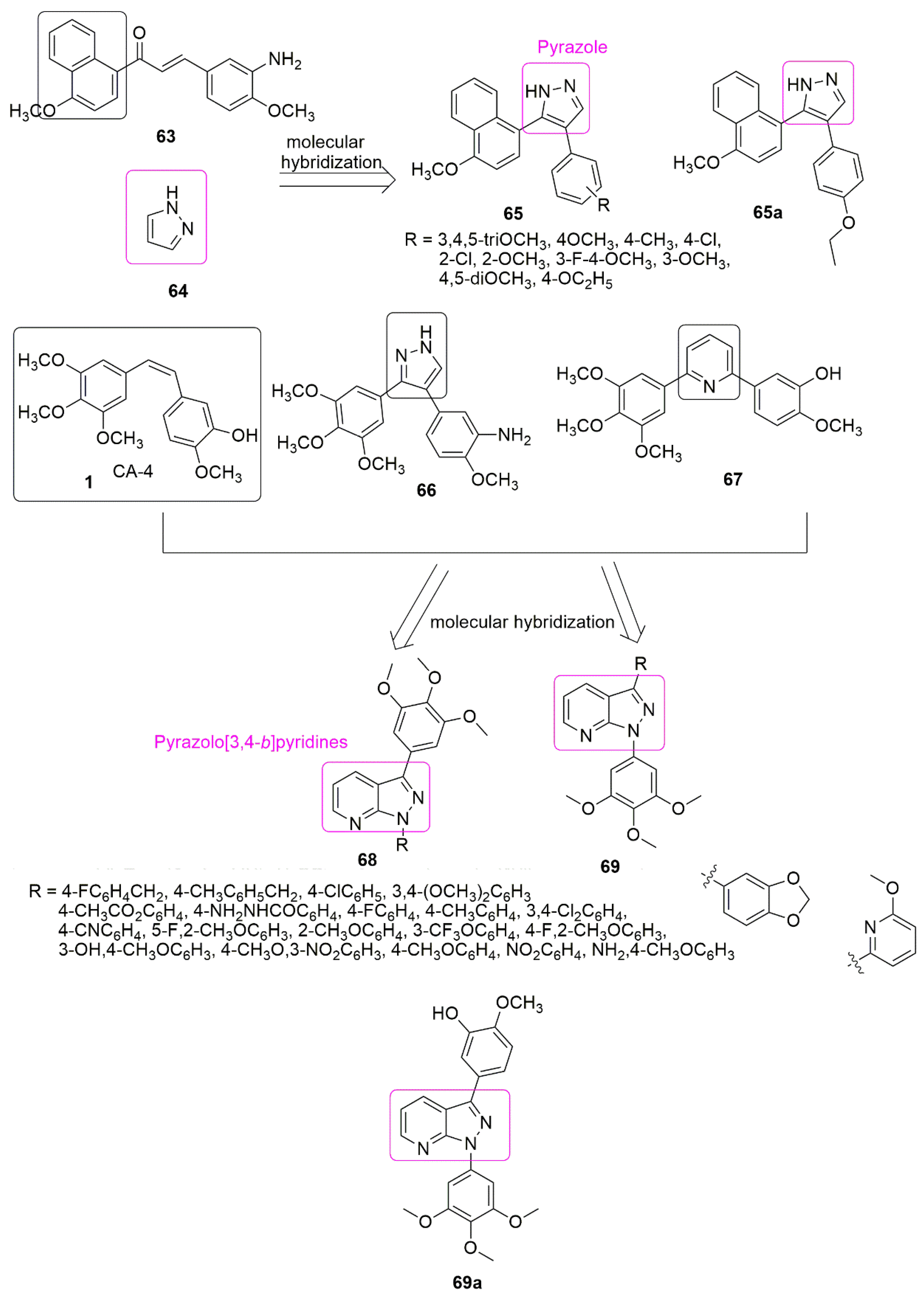
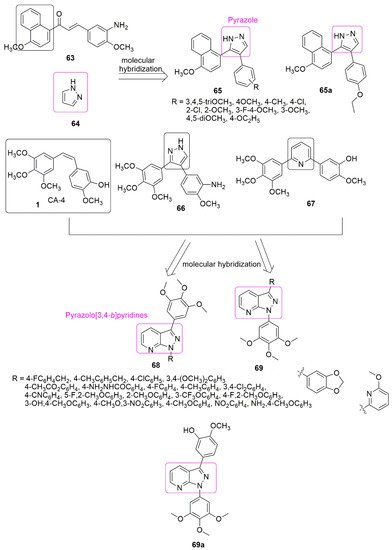
4. Pyrazole Hybrids
Pyrazoles are diazoles with a five-membered ring, useful in organic synthesis, the most studied compounds among the family of azoles. In particular, the heterocyclic cycle has a diverse range of chemical and biological properties [42][43][44] that can be traced back to several well-established therapeutic agents in different classes. Because of the interesting pharmacological properties of pyrazole hybrids, they garnered much attention. The newly synthesized derivatives of pyrazole-naphthalene ( with CF3 group at position two of benzimidazole has the highest potency.
4. Pyrazole Hybrids
Pyrazoles are diazoles with a five-membered ring, useful in organic synthesis, the most studied compounds among the family of azoles. In particular, the heterocyclic cycle has a diverse range of chemical and biological properties [57,58,59] that can be traced back to several well-established therapeutic agents in different classes. Because of the interesting pharmacological properties of pyrazole hybrids, they garnered much attention. The newly synthesized derivatives of pyrazole-naphthalene ( 65) and their in vitro cytotoxicity have been reported [45]. The placement of an electron-donating group at position four of the phenyl ring favors antiproliferative activity. The results of introducing electron-withdrawing groups (R = 4-Cl, 3-Cl, 2-Cl, 3-F, 4-F, 2-F, 3-Br, and 2-Br) into the phenyl ring led to an insignificant increase in the inhibitory activity. Compound ) and their in vitro cytotoxicity have been reported [60]. The placement of an electron-donating group at position four of the phenyl ring favors antiproliferative activity. The results of introducing electron-withdrawing groups (R = 4-Cl, 3-Cl, 2-Cl, 3-F, 4-F, 2-F, 3-Br, and 2-Br) into the phenyl ring led to an insignificant increase in the inhibitory activity. Compound with IC50 = 2.78 ± 0.24 μM bearing ethoxy group (R=4-C2H5O) at the position four of the phenyl ring, was found to be five-fold superior to the standard drug cisplatin (IC50 = 15.24 ± 1.27 μM). Compound
was further identified as a new potent inhibitor of tubulin assembly by inhibiting tubulin polymerization with the IC50 value of 4.6 μM (see
Figure 320. Chemical structures pyrazole hybrids inhibiting tubulin polymerization.
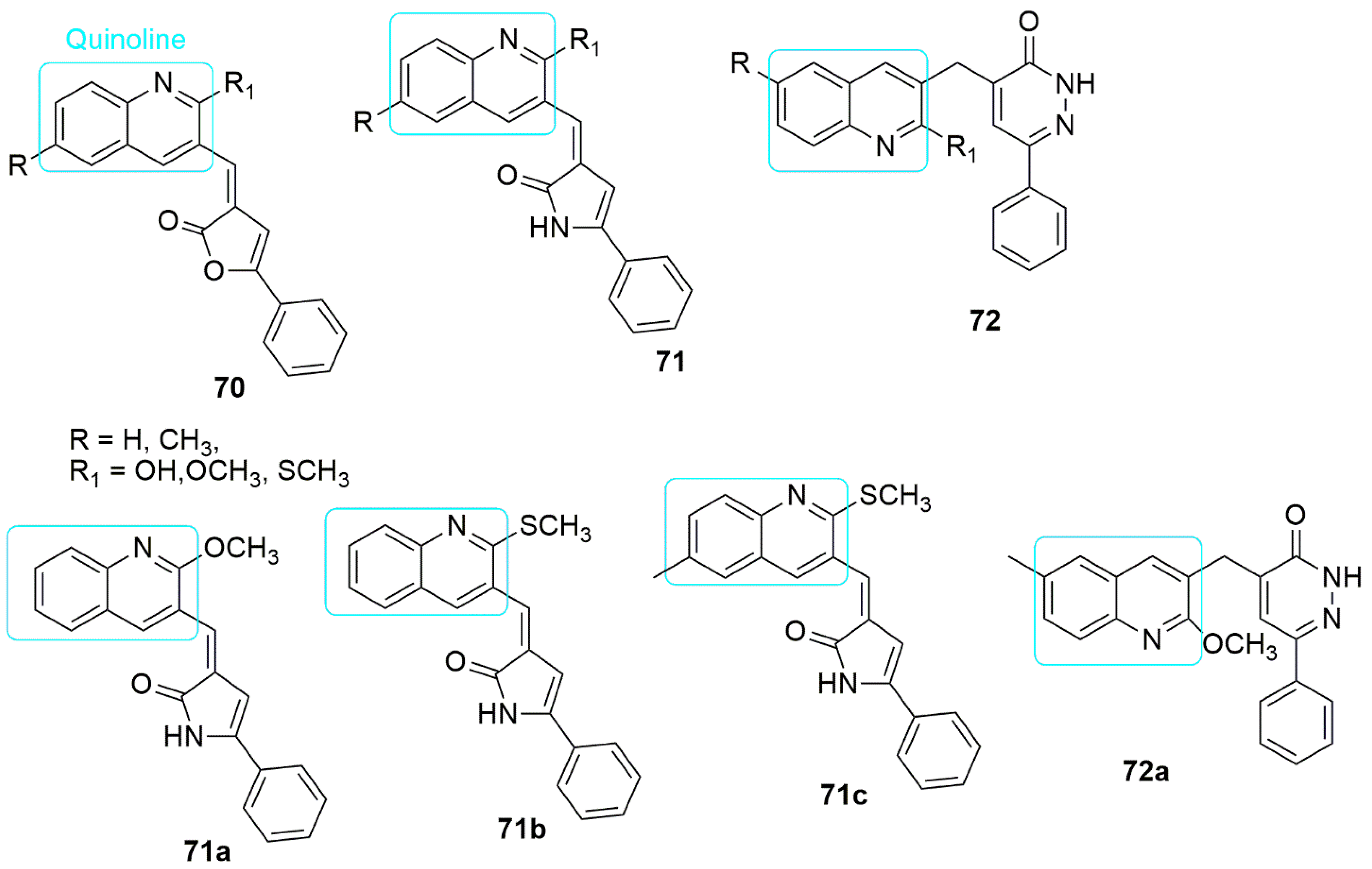
5. Quinoline Hybrids
Pharmacological studies have shown that quinoline’s annulus system has many biological activities. The marketed clinically-approved drugs containing the quinoline core include bedaquiline for multidrug-resistant tuberculosis therapy [46] and irinotecan used to treat colorectal cancer [47]. Quinoline-based compounds play a vital role in the development of anti-cancer drugs. They have been very successful with different mechanisms of action, such as growth inhibition by cell cycle arrest, apoptosis, angiogenesis inhibition, cell migration disruption, and modulation [48].
Quinolinyl pyrrolone compounds can serve as a template for developing antiproliferative agents with tubulin polymerization inhibitory activity. Which can also display pre-G1 apoptosis and cell cycle arrest at the G2/M phase. The visual detail of the molecular docking study showed that Chemical structures pyrazole hybrids inhibiting tubulin polymerization.
5. Quinoline Hybrids
Pharmacological studies have shown that quinoline’s annulus system has many biological activities. The marketed clinically-approved drugs containing the quinoline core include bedaquiline for multidrug-resistant tuberculosis therapy [62] and irinotecan used to treat colorectal cancer [63]. Quinoline-based compounds play a vital role in the development of anti-cancer drugs. They have been very successful with different mechanisms of action, such as growth inhibition by cell cycle arrest, apoptosis, angiogenesis inhibition, cell migration disruption, and modulation [64].
Quinolinyl pyrrolone compounds can serve as a template for developing antiproliferative agents with tubulin polymerization inhibitory activity. Which can also display pre-G1 apoptosis and cell cycle arrest at the G2/M phase. The visual detail of the molecular docking study showed that
binds deep into the hydrophobic pocket and forms interactions with the following residues: Ala250, Leu255, Lys254, Ala316, Ala254, Val318, Leu248, and Ala180, respectively (see
Figure 421. Chemical structures of quinoline conjugates as anti-tubulin agents.
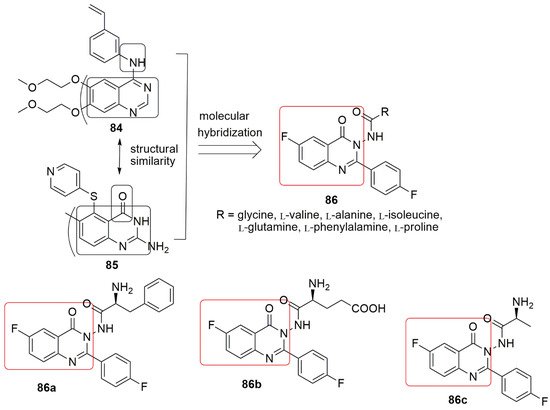
6. Quinazolinone Hybrids
Several quinazolinone derivatives have been synthesized to provide synthetic drugs as more effective medicines for several disorders [49][50][51]. Quinazolinone hybrids constitute a crucial class of compounds with diverse therapeutic properties due to various substitutions on the ring system. Importantly, some quinazolinone analogs exhibited excellent anticancer activity via inhibition of dihydrofolate reductase enzyme [52]. Quinazolinones have also served as anti-tumor agents and inhibitors of tubulin polymerization [53]. Zayed and co-workers reported new hybrid molecules containing quinazolinone nucleus merged with various forms of L-amino acids at position three by exploring the structure of erlotinib Chemical structures of quinoline conjugates as anti-tubulin agents.
6. Quinazolinone Hybrids
Several quinazolinone derivatives have been synthesized to provide synthetic drugs as more effective medicines for several disorders [69,70,71]. Quinazolinone hybrids constitute a crucial class of compounds with diverse therapeutic properties due to various substitutions on the ring system. Importantly, some quinazolinone analogs exhibited excellent anticancer activity via inhibition of dihydrofolate reductase enzyme [72]. Quinazolinones have also served as anti-tumor agents and inhibitors of tubulin polymerization [73]. Zayed and co-workers reported new hybrid molecules containing quinazolinone nucleus merged with various forms of L-amino acids at position three by exploring the structure of erlotinib 85 [54]. The compounds were exposed to cytotoxic screening using two breast cancer cell lines, mainly MCF-7 and MDA-MB-231. All the synthesized compounds showed good activity against the MCF-7 cell line. Compound [74]. The compounds were exposed to cytotoxic screening using two breast cancer cell lines, mainly MCF-7 and MDA-MB-231. All the synthesized compounds showed good activity against the MCF-7 cell line. Compound displays better activity than the reference erlotinib against the MCF-7 cell line. The most active compound,
(IC50 = 0.44 ± 0.01 µM), against MCF-7, contains an L-phenylalanine substitution at position 3 of the quinazolinone scaffold. In comparison,
(IC50 = 0.43 ± 0.02 µM), the most active compound against the MDA-MBA-231 cell line, has L-glutamine at position 3 of quinazolinone. Compound
was the least active derivative for both cell lines. The tubulin polymerization assay revealed
as a potent tubulin inhibitor (IC50 = 6.24 µM), which may explain these compounds’ high cytotoxic activity. The in silico study detailed that
occupied the same landscape in the colchicine binding site with the formation of a strong hydrogen bond between the compounds and tubulin residues Tyrα224, GlnA111, a Glnβ247, and Leuβ248, respectively (see
Figure 25. Chemical structures of quinazolinones derivatives evaluated for biological activity.
7. Quinolinone Hybrids
Heterocycle-fused quinolinone is another privileged structure used as a building block in drug discovery. The quinolinone hybrids have shown biological activities, including antimicrobial [55], anti-inflammatory [56], anticancer [57], anti-Alzheimer [58], and anti-leishmanial [59] effects. Interestingly, quinolinone derivatives can successfully inhibit IDH1 mutants R132H, R132C, R132G, and R132L and display good selectivity vs. the wild-type IDH protein [60]. In addition, 7-phenyl-pyrroloquinolinones (7-PPyQs) and, mainly, the 3-substituted derivatives bind to the colchicine site with high affinity. Obstructing microtubule assembly and thus engendering excellent antiproliferative effect. This makes the compound have similar inhibitory activities to CA-4. It was believed that the [3,2-f] configuration, the 7-phenyl, and 9-carbonyl groups, and the ethyl group at position 3 would be crucial for suitable anti-tubulin activities. Considering these facts, Bortolzzi and colleagues designed synthesized hybrids mimicking PPyQ [61]. Compounds Chemical structures of quinazolinones derivatives evaluated for biological activity.
7. Quinolinone Hybrids
Heterocycle-fused quinolinone is another privileged structure used as a building block in drug discovery. The quinolinone hybrids have shown biological activities, including antimicrobial [75], anti-inflammatory [76], anticancer [77], anti-Alzheimer [78], and anti-leishmanial [79] effects. Interestingly, quinolinone derivatives can successfully inhibit IDH1 mutants R132H, R132C, R132G, and R132L and display good selectivity vs. the wild-type IDH protein [80]. In addition, 7-phenyl-pyrroloquinolinones (7-PPyQs) and, mainly, the 3-substituted derivatives bind to the colchicine site with high affinity. Obstructing microtubule assembly and thus engendering excellent antiproliferative effect. This makes the compound have similar inhibitory activities to CA-4. It was believed that the [3,2-f] configuration, the 7-phenyl, and 9-carbonyl groups, and the ethyl group at position 3 would be crucial for suitable anti-tubulin activities. Considering these facts, Bortolzzi and colleagues designed synthesized hybrids mimicking PPyQ [81]. Compounds 88c emerged as the most active against a panel of seven human tumor cell lines including human T-cell leukemia (Jurkat and CEM), human B-cell leukemia (RS4; 11), human myeloid leukemia (Kasumi-1), breast (MDA-MB-231), cervix (HeLa), lung (A549) and colon (HT-29) cell lines as reported in the work of Bortolzzi [61]. The replacement of the ethyl group of emerged as the most active against a panel of seven human tumor cell lines including human T-cell leukemia (Jurkat and CEM), human B-cell leukemia (RS4; 11), human myeloid leukemia (Kasumi-1), breast (MDA-MB-231), cervix (HeLa), lung (A549) and colon (HT-29) cell lines as reported in the work of Bortolzzi [81]. The replacement of the ethyl group of with a bulky linear alkyl chain (18 C) such as (R = CH2)17CH3, R1 = CH3) and (R = CH2)17CH3, R1 = phenyl) caused a drastic loss of antiproliferative activity in all cell lines. The SAR evaluation indicated that the increase in the alkyl chain attached to 3N decreased cytotoxicity. Compounds
displayed moderate to poor activity, while
, a diazepine-indole derivative, displayed poor activity (GI50s > 10,000 nM). Moreover,
inhibited tubulin assembly with IC50 values of 0.99, 1.1, and 0.84 µM, respectively, which was higher than the IC50 values of the reference compounds (CA-4, IC50 = 0.64 µM).
8. Chalcone Hybrids
Chalcones are open-chain compounds with a chemical framework of 1,3-diaryl-2-propen-1-one, also known as chalconoid, which occurs as trans and cis isomers, with two aromatic rings linked to three-carbon α,β-unsaturated carbonyl. The thermodynamical stability of the trans isomer has made it the leading configuration among the chalcones. In contrast, the configuration of the cis isomer is thermodynamically unpredictable due to the sturdy steric influences between the carbonyl group and one of the aromatic rings.
In the eagerness of synergism in terms of activity, with enhanced affinity and efficacy about the parent structural components, Sultana and colleagues designed some chalcone conjugates by replacing the benzene ring of the chalcone with benzo[d]imidazo[2,1-b]thiazole in the YM-201627
93 structural compound [62]. Meanwhile, structural compound [88]. Meanwhile, 93 has been reported as an effective and orally active antitumor agent [63]. Various substituents on both the pharmacophores were explored, and 27 compounds were synthesized and evaluated for biological activities. In addition, four panels of human cancer cell lines, mainly lung (A-549), breast (MDA MB-231), prostate (DU-145), and colon cancer (HT-29), were used in the MTT assay. All the compounds exhibited moderate to good antiproliferative activity with IC50 values ranging from 1.28 to 50 µM. Among the derivatives, the most active conjugates were has been reported as an effective and orally active antitumor agent [89]. Various substituents on both the pharmacophores were explored, and 27 compounds were synthesized and evaluated for biological activities by the authors. In addition, four panels of human cancer cell lines, mainly lung (A-549), breast (MDA MB-231), prostate (DU-145), and colon cancer (HT-29), were used in the MTT assay. All the compounds exhibited moderate to good antiproliferative activity with IC50 values ranging from 1.28 to 50 µM. Among the derivatives, the most active conjugates were (R = OCH3, R1 = R4 = H, R2 = OH, R3 = OCH3) and
95b (R = R1= R4 = R2 = H, R3 = OCH3) with IC50 values 1.3 and 1.2 µM against MDA MB-231. The order of potency in regard to the substituents on ring D is 4-OCH3 > 3-OH- 4- OCH3 > 3, 4-diOCH3 > 3, 4, 5-triOCH3 > 2-Br-3,4,5-tri OCH3 > 3, 5-diOCH3. The anti-tubulin evaluation of the two compounds showed better IC50 values (1.93 and 1.88 µM) than nocodazole (IC50 =1.97 µM).
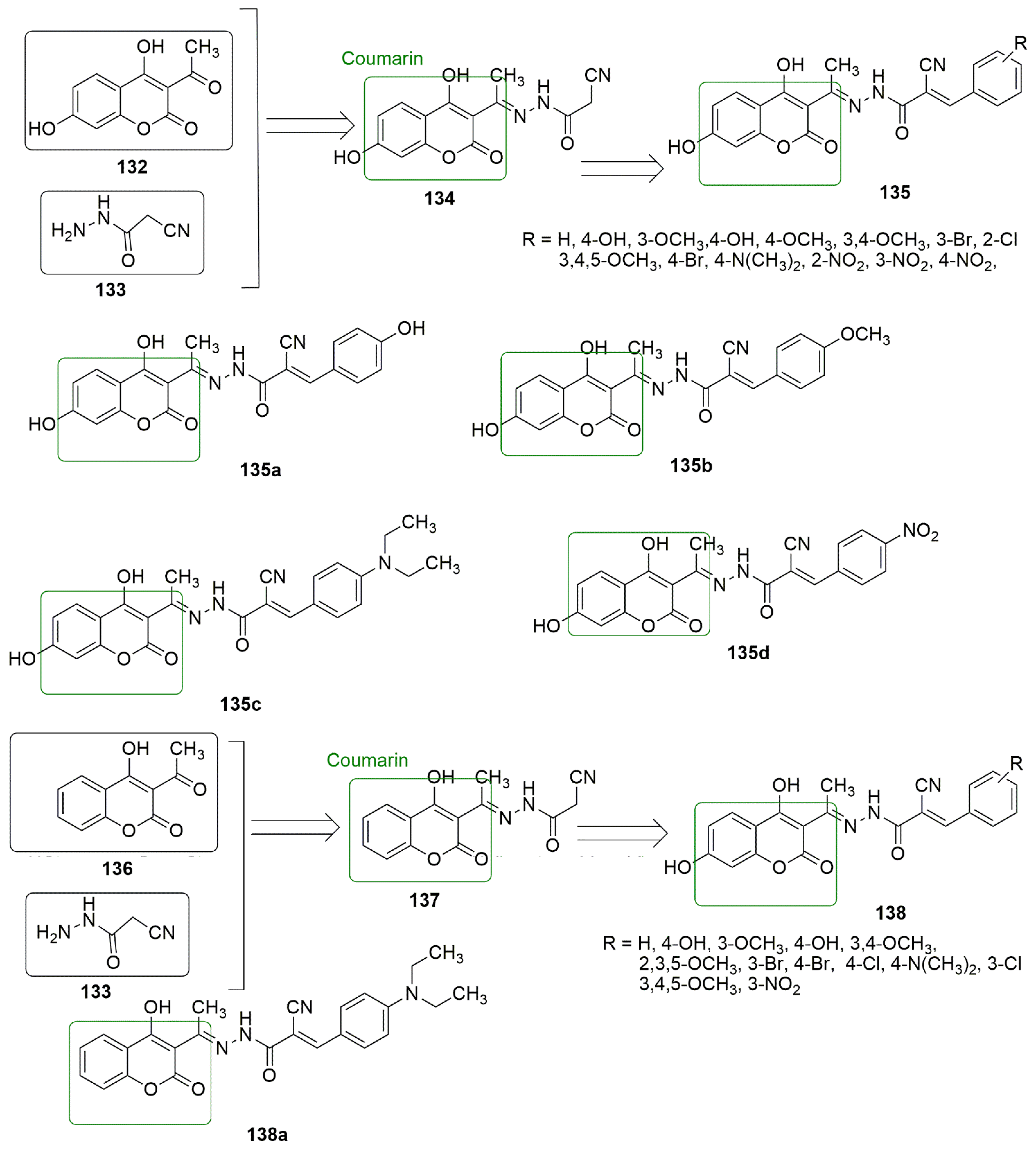
9. Coumarin Hybrids
Coumarin is also known as 2H-1-benzopyran-2-one, benzo-α-pyrone, phenylpropanoids, cis-o-coumarinic acid lactone, coumarinic anhydride, and 5,6-benzo-2-pyrone. They are secondary metabolites of plants, bacteria, fungi, and sponges. Physically, coumarin is a crystalline colorless solid with a specific sweet odor. Coumarin has been classified as an oxygenated heterocycle rather than a benzoic acid derivative. Many coumarin hybrids have been reported to be used for medical disorders, namely as anti-allergic [64], antiviral [65], vasodilatory [66], antibacterial [67][68], exhibiting cyclooxygenase inhibition [69], lipoxygenase [70], antithrombotic [71], and anticancer activity [72]. The molecular hybridization of the coumarin nucleus to a biodynamic system that is a biologically active pharmacophore has enhanced biological activities. These compounds with heterocycle-coumarin’s broad pharmacological activity have gained momentous attention in medicinal chemistry. Coumarin has also been reported to possess many pharmacological activities, such as tubulin polymerization inhibition. In the view of coumarin (R = R1= R4 = R2 = H, R3 = OCH3) with IC50 values 1.3 and 1.2 µM against MDA MB-231. The order of potency in regard to the substituents on ring D is 4-OCH3 > 3-OH- 4- OCH3 > 3, 4-diOCH3 > 3, 4, 5-triOCH3 > 2-Br-3,4,5-tri OCH3 > 3, 5-diOCH3. The anti-tubulin evaluation of the two compounds showed better IC50 values (1.93 and 1.88 µM) than nocodazole (IC50 =1.97 µM).
9. Coumarin Hybrids
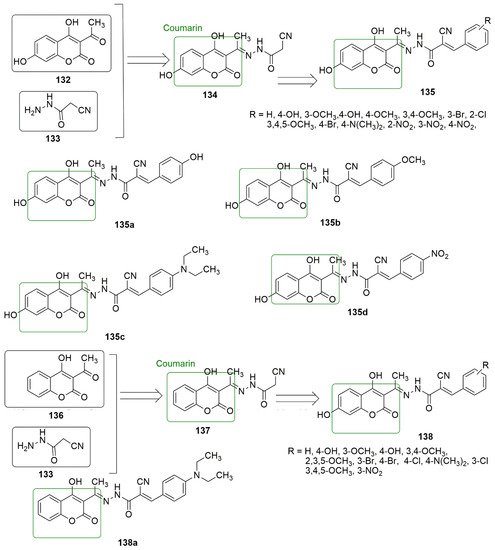
Coumarin is also known as 2H-1-benzopyran-2-one, benzo-α-pyrone, phenylpropanoids, cis-o-coumarinic acid lactone, coumarinic anhydride, and 5,6-benzo-2-pyrone. They are secondary metabolites of plants, bacteria, fungi, and sponges. Physically, coumarin is a crystalline colorless solid with a specific sweet odor. Coumarin has been classified as an oxygenated heterocycle rather than a benzoic acid derivative. Many coumarin hybrids have been reported to be used for medical disorders, namely as anti-allergic [103], antiviral [104], vasodilatory [105], antibacterial [106,107], exhibiting cyclooxygenase inhibition [108], lipoxygenase [109], antithrombotic [110], and anticancer activity [111]. The molecular hybridization of the coumarin nucleus to a biodynamic system that is a biologically active pharmacophore has enhanced biological activities. These compounds with heterocycle-coumarin’s broad pharmacological activity have gained momentous attention in medicinal chemistry. Coumarin has also been reported to possess many pharmacological activities, such as tubulin polymerization inhibition. In the view of coumarin 133 as cytotoxic agents, Govindaiah and colleagues reported for the first time the amalgamation and cytotoxicity activity of 4′,7-dihydroxycoumarin bearing cyanohydrazone moieties [73]. A series of 4,7-dihydroxycoumarin based acryloylcyanohydrazone hybrids as cytotoxic agents, Govindaiah and colleagues reported for the first time the amalgamation and cytotoxicity activity of 4′,7-dihydroxycoumarin bearing cyanohydrazone moieties [112]. A series of 4,7-dihydroxycoumarin based acryloylcyanohydrazone hybrids was synthesized and evaluated for antiproliferative activity against a panel of four cancer cell lines, namely A549, HeLa, SKNSH (neuroblastoma), and MCF7. Among the tested compounds,
showed notable antiproliferative activity with IC50 values ranging from 3.42 to 10.26 μM against all the tested cancer cell lines.
135c (R = 4-N(CH3)2) emerged as the most active compound with IC50 values of 4.31 ± 0.04, 5.14 ± 0.16, 6.09 ± 0.32, and 3.42 ± 0.52 μM against A549, HeLa, SKNSH, and MCF7, respectively. The SAR detailed that substitution at the para position (R = 4-OCH3, 4-Br, 4-N(CH3)2 and 4-NO2) and ortho position compounds (R = 2-Cl and 4-NO2) increases the anticancer activity compared to substitution at the meta position compounds (3-NO2 and 3-Br). The substitution of the electron-donating group at the para position enhanced the inhibitory activity more than the electron-withdrawing group. The pharmacological mechanistic studies of
(R = 4-N(CH3)2) emerged as the most active compound with IC50 values of 4.31 ± 0.04, 5.14 ± 0.16, 6.09 ± 0.32, and 3.42 ± 0.52 μM against A549, HeLa, SKNSH, and MCF7, respectively. The SAR detailed that substitution at the para position (R = 4-OCH3, 4-Br, 4-N(CH3)2 and 4-NO2) and ortho position compounds (R = 2-Cl and 4-NO2) increases the anticancer activity compared to substitution at the meta position compounds (3-NO2 and 3-Br). The substitution of the electron-donating group at the para position enhanced the inhibitory activity more than the electron-withdrawing group. The authors further evaluated the pharmacological mechanistic studies of
135c on cell cycle progression were further evaluated and carried out a tubulin polymerization inhibition assay. The results indicate that
on cell cycle progression and carried out a tubulin polymerization inhibition assay. The results indicate that
arrests the cell cycle at the G2/M phase and inhibits tubulin polymerization with an IC50 value of 6.19 μM; thus, the compound can be a candidate antimitotic agent for treating cancer by targeting tubulin protein. The docking studies revealed that hydrazide–hydrazone backbone is more active when the cyano group is attached to the hydrazone moiety: This improved the activity of the compound (see
Figure 637. Novel coumarin-based acryloylcyanohydrazone compounds evaluated against a different panel of cancer cell lines.
10. Indole Hybrids
Indole is also known as benzopyrrole, a bicyclic heterocyclic molecule with a benzenoid system. It has ten π-electrons, lone pairs from nitrogen, and four double bonds that provide eight valence electrons. When an electrophilic substitution occurs at C-3 of indole moiety, it becomes more reactive than benzene. Indole scaffolds have the exceptional characteristics of imitating the structure of peptides and binding in a reversible way to enzymes that offer enormous possibilities for discovering new medicines with different modes of action [74].
Many indole-based compounds have been reported as tubulin polymerization inhibitors binding to the colchicine domain, including IPP51 ( Novel coumarin-based acryloylcyanohydrazone compounds evaluated against a different panel of cancer cell lines.
10. Indole Hybrids
Indole is also known as benzopyrrole, a bicyclic heterocyclic molecule with a benzenoid system. It has ten π-electrons, lone pairs from nitrogen, and four double bonds that provide eight valence electrons. When an electrophilic substitution occurs at C-3 of indole moiety, it becomes more reactive than benzene. Indole scaffolds have the exceptional characteristics of imitating the structure of peptides and binding in a reversible way to enzymes that offer enormous possibilities for discovering new medicines with different modes of action [114].
Many indole-based compounds have been reported as tubulin polymerization inhibitors binding to the colchicine domain, including IPP51 (
142). IPP51 is selective toward the proliferation of bladder carcinoma cells and vies with colchicine for binding to the tubulin [75]. While the trimethoxy analog ). IPP51 is selective toward the proliferation of bladder carcinoma cells and vies with colchicine for binding to the tubulin [124]. While the trimethoxy analog 142 showed antiproliferative activity against four human and a murine glioblastoma cell line, interacted with tubulin, and fitted into the colchicine binding site [76]. Meanwhile, MX58151 ( showed antiproliferative activity against four human and a murine glioblastoma cell line, interacted with tubulin, and fitted into the colchicine binding site [125]. Meanwhile, MX58151 ( 144), bearing a 3-bromo-4,5- dimethoxy-phenyl moiety, are new microtubule inhibitors [77][78]. Based on the usefulness of 3-bromo-4,5-dimethoxy-group and indole moiety in antiproliferative and tubulin polymerization inhibitory effectiveness, Mirzaei and colleagues combined chalcone and indole scaffolds into a single entity, which are indole-based chalconoids [79]. The amalgamation of indole-chalcone scaffolds ( ), bearing a 3-bromo-4,5- dimethoxy-phenyl moiety, are new microtubule inhibitors [126,127]. Based on the usefulness of 3-bromo-4,5-dimethoxy-group and indole moiety in antiproliferative and tubulin polymerization inhibitory effectiveness, Mirzaei and colleagues combined chalcone and indole scaffolds into a single entity, which are indole-based chalconoids [128]. The amalgamation of indole-chalcone scaffolds ( ) and in vitro biological evaluation as tubulin-targeting anti-cancer agents were reported. The in vitro antiproliferative activity of the synthesized compounds were examined against adenocarcinoma human alveolar basal epithelial cells (A549), breast cancer cells (MCF7), and human ovarian carcinoma cells (SKOV3) using an MTT assay. (E)-stereoisomer was assigned for all the synthesized compounds because the detected coupling constants (J) between vinylic hydrogens were more significant (J = 15 Hz). Among the synthesized compounds,
(IC50 value = 4.3 ± 0.2 µM) showed superior cytotoxicity against human alveolar basal epithelial (A549) and SKOV3 cells, while
was more potent against the MCF7 cell line (11.7 ± 0.2 µM). In addition,
inhibited tubulin polymerization (IC50 values = 17.8 ± 0.2, 18.3 ± 0.4, and 40.0 ± 0.1 µM, respectively). Compound
reduced the mitochondrial thiol content in a concentration-dependent manner. Cytotoxic effects of
145a can be possibly correlated to tubulin polymerization inhibition, reactivity toward mitochondrial thiols, and induction of mitochondrial pathway of apoptosis.
11. Oxindole Hybrids
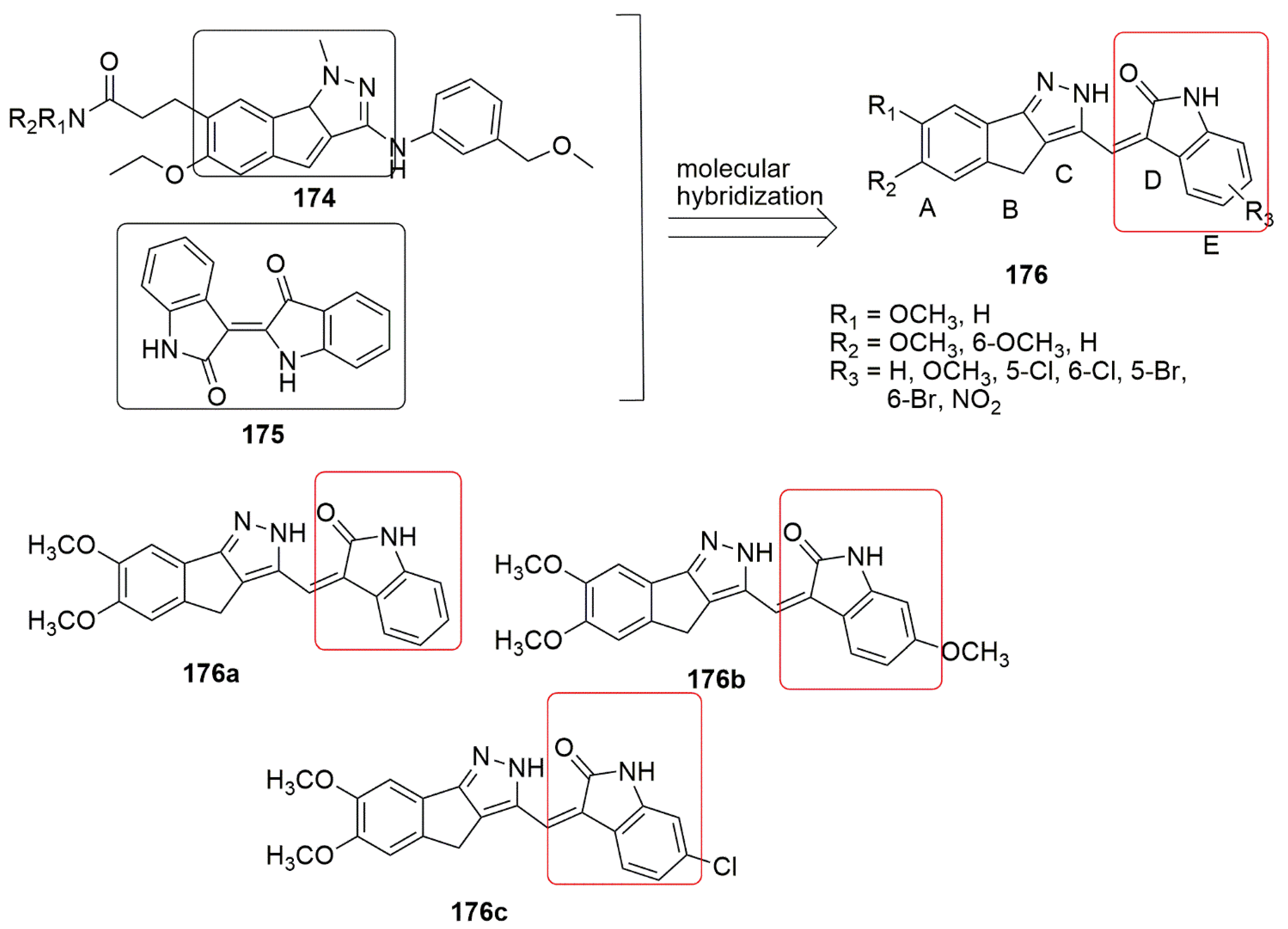
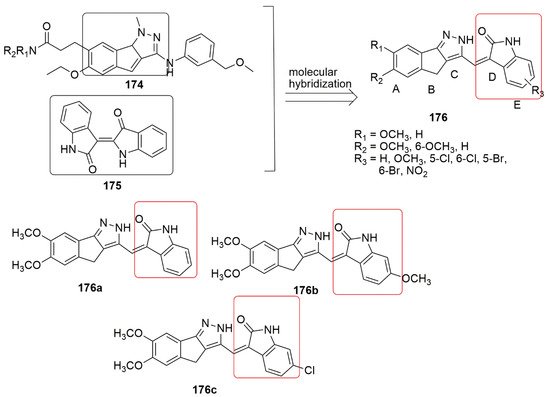
Many medicinal chemistry researchers have shown enormous interest in developing novel oxindole hybrids due to their invaluable biological potential. Marketed oxindole derivatives include sunitinib and indolidan. Sunitinib is used to treat gastrointestinal stromal tumors and advanced renal cell cancer [80], while indolidan is a phosphodiesterase inhibitor [81][82]. Other oxindole derivatives, such as SU5416, SU5402, and S 6668, have exhibited potent antitumor activity and are known as receptor tyrosine kinase (RTK) inhibitors [82][83]. 1,4-Dihydroindeno[1,2-c]pyrazole ( can be possibly correlated to tubulin polymerization inhibition, reactivity toward mitochondrial thiols, and induction of mitochondrial pathway of apoptosis.
11. Oxindole Hybrids
Many medicinal chemistry researchers have shown enormous interest in developing novel oxindole hybrids due to their invaluable biological potential. Marketed oxindole derivatives include sunitinib and indolidan. Sunitinib is used to treat gastrointestinal stromal tumors and advanced renal cell cancer [145], while indolidan is a phosphodiesterase inhibitor [146,147]. Other oxindole derivatives, such as SU5416, SU5402, and S 6668, have exhibited potent antitumor activity and are known as receptor tyrosine kinase (RTK) inhibitors [147,148]. 1,4-Dihydroindeno[1,2-c]pyrazole ( 174) has acted as an inhibitor such as cyclin-dependent kinase (CDK), PDGFR tyrosine kinase inhibitor, checkpoint kinase 1 (Chk1) inhibitors, tyrosine kinases (EGFR and VGEFR-2), hypoxia-inducible factor receptor (HIF-1) [84]. A novel series of 1, 4-dihydroindeno-[1,2-c] pyrazole in combination with the oxindole ( ) has acted as an inhibitor such as cyclin-dependent kinase (CDK), PDGFR tyrosine kinase inhibitor, checkpoint kinase 1 (Chk1) inhibitors, tyrosine kinases (EGFR and VGEFR-2), hypoxia-inducible factor receptor (HIF-1) [149]. A novel series of 1, 4-dihydroindeno-[1,2-c] pyrazole in combination with the oxindole ( 175) scaffold have been synthesized by Khan and co-workers [84]. The compounds were synthesized using the Knoevenagel condensation approach and further evaluated for their antiproliferative activity against four panels of cancer cell lines, namely, HeLa, A549, and MDA-MB-231 human cancer cell lines as well as HEK-293 (normal human embryonic kidney cells). Among the tested compounds, ) scaffold have been synthesized by Khan and co-workers [149]. The compounds were synthesized using the Knoevenagel condensation approach and further evaluated for their antiproliferative activity against four panels of cancer cell lines, namely, HeLa, A549, and MDA-MB-231 human cancer cell lines as well as HEK-293 (normal human embryonic kidney cells). Among the tested compounds, , superior cytotoxicity with IC50 values varying from 1.33 to 4.33 µM. Meanwhile, the tubulin polymerization assay demonstrated that
triggers microtubule assembly and boosts the G2/M checkpoint proteins (Cyclin B1 and CDK1), whereas compound
induces apoptosis through the activation of caspase-3 besides. Importantly, the presence of a Cl or OCH3 substituent at ring E was found to be crucial for excellent anti-tubulin activity (see
Figure 746. 1, 4-dihydroindeno-[1,2-c] pyrazole in combination with oxindole.
12. Conclusions
The concept of designing hybrid molecules containing two or more chemical entities a single molecule is now used in drug discovery. Many medicinal chemistry researchers presently use the molecular hybridization approach in developing unique bioactive compounds acting as dual inhibitors, while being less susceptible to drug resistance. The molecular amalgamation involved the pharmacophoric unit of a known protypes [85] which are significantly active to produce a single entity. This approach was utilized (i) for modulation of objectionable secondary effects and (ii) to identify dual-acting molecule capable of reproducing the effect of combinations of more than one therapeutic agent. Triazole, tetrazole, benzimidazole, pyrazole, quinoline, quinazolinone, quinolinone, chalcone, coumarin, indole, and oxindole are attractive molecules with potent anticancer activities that have gained more attention thus they are used as building blocks by many researchers for designing and developing novel chemical structures.
1, 4-dihydroindeno-[1,2-c] pyrazole in combination with oxindole.
12. Conclusions
The concept of designing hybrid molecules containing two or more chemical entities a single molecule is now used in drug discovery. Many medicinal chemistry researchers presently use the molecular hybridization approach in developing unique bioactive compounds acting as dual inhibitors, while being less susceptible to drug resistance. The molecular amalgamation involved the pharmacophoric unit of a known protypes [151] which are significantly active to produce a single entity. This approach was utilized (i) for modulation of objectionable secondary effects and (ii) to identify dual-acting molecule capable of reproducing the effect of combinations of more than one therapeutic agent. Triazole, tetrazole, benzimidazole, pyrazole, quinoline, quinazolinone, quinolinone, chalcone, coumarin, indole, and oxindole are attractive molecules with potent anticancer activities that have gained more attention thus they are used as building blocks by many researchers for designing and developing novel chemical structures.