Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Conner Chen and Version 2 by Conner Chen.
Evolutionary medicine is a rapidly developing field. There are two polyploidy-related aspects of it. Firstly, mutations and changes in expression patterns of many ohnologs are associated with various diseases and developmental disorders. Secondly (and probably more important), cancer is now interpreted as an evolutionary phenomenon, and polyploidy often arises in cancer.
- evolutionary medicine
1. Evolutionary Medicine
Evolutionary medicine is a rapidly developing field [1]. There are two polyploidy-related aspects of it. Firstly, mutations and changes in expression patterns of many ohnologs are associated with various diseases and developmental disorders [2]. Secondly (and probably more important), cancer is now interpreted as an evolutionary phenomenon, and polyploidy often arises in cancer [3][4][5].
2. Ohnologs and Diseases
The currently existing ohnologs are in gene balance as dosage-sensitive genes [6][7][8]. Gene-dosage sensitivity has been of increasing interest as it might provide a clue for many pathologies [8]. The gene balance model states that a stoichiometric equilibrium is maintained among all of the complex gene products in a pathway, so a change in expression, mutation or copy number variation in a single gene of a pathway would be deleterious. The changes in ohnolog expression patterns are found in different types of cancers (breast, prostate, colon, thyroid, ovary, bladder, cervix, lung, uterus) and involved in tumor angiogenesis [7]. Human monogenic disease genes in the OMIM database are enriched in ohnologs [8]. Ohnologs are also highly enriched in the genes from the Disease Gene Network and the Network of Cancer Genes, which involve diseases with more complex etiologies (Figure 1). The NCG were from [9], the DGN (curated part) were from [10].
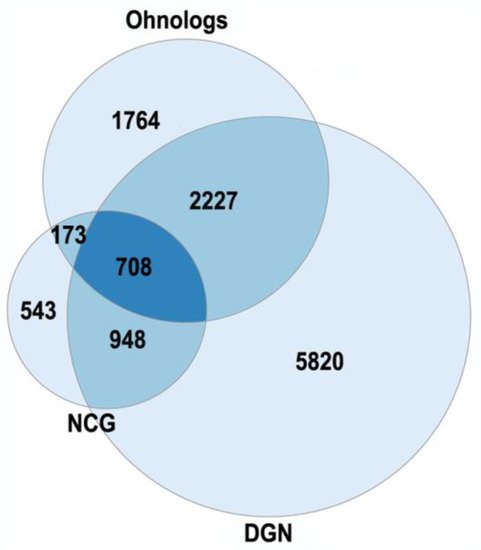
Figure 1. The enrichment of human ohnologs in Network of Cancer Genes (NCG) and Disease Gene Network (DGN). NCG: p < 10−46, DGN: p < 10−107.
3. Polyploidy in Cancer
The main contradiction of multicellularity stems from its evolutionary origin, as it is between the cellular and organismal levels [11]. Cell pluripotency and proliferative potential are vital for the healthy development and longevity of multicellulars if held in check. In this case, activity at the unicellular level promotes an organism’s vitality. In contrast, runaway unicellularity results in cancer when cells tend to behave as independent evolutionary units. The atavistic theory of oncogenesis assumes that cancer is a reversal from a multicellular to a unicellular state and is frequently associated with polyploidization [5][12][13]. The genes and transcriptome modules of unicellular origin are overexpressed in human cancers, whereas multicellular genes and modules are downregulated [4][14].
These results were obtained on the bulk tissues and without control for cell cycle activity. Therefore the picture remained uncertain as: (i) cell cycle activity can be higher in cancer tissues; and (ii) expression of unicellular genes positively correlates with expression of cell cycle genes even if the overlapping genes are removed [15]. Yet, the upregulation of unicellular genes and the unicellular giant cluster of interactome was recently observed in the single-cell transcriptomes of various cancer types with the control for cell cycle activity [15]. Furthermore, the upregulation of unicellular genes and interactome giant cluster was shown in the invasive myeloma cells as compared with non-invasive cells. The upregulation of the unicellular giant cluster suggests that oncogenesis is not just an alteration in a few genes but the switching to ancient unicellular programs (when cells tend to behave as independent organisms). The interactome clusters have a higher interaction density within than between them, therefore they can serve as attractors (stable states of dynamic systems) of cellular programs. The unicellular cluster is denser (it has a higher inside/outside interaction ratio compared with multicellular clusters), which indicates a stronger attractor effect and helps explain why cells of multicellular organisms are prone to oncogenesis [11]. These observations suggest practical applications as certain unicellular-specific drugs can be applied for the treatment of cancer [16].
The upregulation of G2/M checkpoint genes with downregulation of G1/S checkpoint was found in polyploid breast cancer cells (called ‘G2/M vs. G1/S checkpoint inversion’) using single-cell transcriptomes with the control for cell cycle activity [15]. Probably, the hyper-activation of the G2/M checkpoint (which can be caused by DNA damage) retards the G2/M transition. In accordance with this suggestion, the genes belonging to G2/M transition were downregulated in polyploid cells, whereas the genes belonging to G1/S transition were upregulated (‘inversion of G2/M vs. G1/S transition’). The cytokinesis-related genes were downregulated [15].
Polyploidy results from the overall instability of metabolically stressed cancer cells [5][17]. In single-cell transcriptomes, the cancer cells show a higher scatter around the regression line of expression of the unicellular genes and interactome giant cluster on the cell cycle genes compared with normal cells [15]. This observation demonstrates a greater variability of cancer cells caused by their deviation from a stable, ‘canalized’ line (sensu [18]) of normal cells. Polyploidy facilitates chromosomal instability, which leads to aneuploidy and chromosome abnormalities increasing the probability of transformation [5][19]. Notably, under the influence of a carcinogenesis promoter (12-O-tetradecanoylphorbol-13-acetate), human lymphocytes in primary culture continue DNA synthesis even when mitosis is blocked by colchicine or cytokinesis is blocked by cytochalasine, thereby forming cells with high ploidy [20]. This indicates the role of the loosening of cell cycle control in pathological polyploidization. Polyploidy in tumors is associated with resistance to radio/chemotherapy and poor prognosis [21][22].
Intriguingly, polyploidization by fusion of different cell types (epigenetic hybridization), which is frequently observed both in normal and cancer tissues [23][19], strikingly resembles allopolyploidization (genetic hybridization) in the evolution and agri/aquaculture described above. Cell fusion plays a crucial role in several physiological processes, including wound healing and tissue regeneration [23]. Thus, the bone marrow-derived stem cells (BMSCs) could adopt the specific properties of a different organ by cell fusion, thereby restoring organ function [23]. Similar to the allopolyploid organisms, the hybrid polyploid cells acquire stress resistance (e.g., chemo- and radiation-resistance) and adaptive plasticity. For instance, fusion with motile leucocytes such as macrophages renders cancer cells an ability to migrate, which plays a major role in metastasis [19]. Furthermore, cell fusion can be a mechanism of cancer stem cell formation [19]. Cancer stem cells present an important problem as they are resistant to therapy and serve as a source of new cells after the reduction in tumor mass by treatment [19][24].
Thus, polyploidy in cancer is associated with a poor prognosis as a result of resistance to stress, stemness, and an enhanced migration ability [5][19]. Paradoxically, albeit in normal tissues polyploidization frequently accompanies differentiation [25][26], polyploidy in tumors is associated with the features of fetality and stemness [5][19]. This paradox can be solved if considering cancer from an evolutionary viewpoint and comparing it with organismal polyploidy. Polyploid cancer cells show a general increase in adaptivity, which is reminiscent of the rapid growth, stress resistance, and the evolutionary plasticity of polyploid organisms. Stress caused by diseases, which results in the formation and survival of polyploid cells, can be considered as an analog of environmental stress conferring an adaptive advantage to polyploid organisms. In the polyploid cancer cells, the expression of the unicellular giant cluster of interactome is upregulated, whereas the multicellular cluster is downregulated, even as compared with diploid cells from the same cancer (Figure 2A,B). The pluripotency signature (PluriNet) is upregulated, whereas the genes involved in the regulation of multicellular organismal development are downregulated (Figure 2C,D). These facts indicate that the polyploidization of cancer cells enhances their unicellular properties and proliferative potential, at the same time reducing the multicellular-level control. The up- or down-regulated genes in polyploid cancer cells were from [27], which was based on the analysis of about ten thousand cancer samples. The UC and MC cluster genes were from [11], the PluriNet genes were from MSigDB [28].
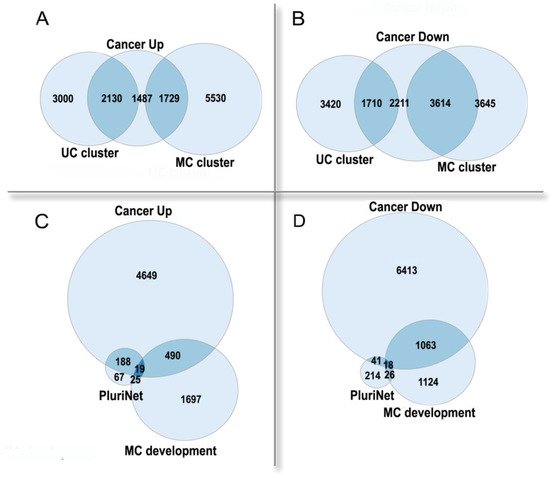
Figure 2. The enrichment of human genes, which are up- or down-regulated in polyploid cancer cells as compared with diploid cells of the same cancer, in functional gene groups. (A,B)—unicellular (UC) and multicellular (MC) giant clusters of interactome.UC cluster: enrichment p < 10−157 in Up, underrepresentation p < 10−15 in Down.MC cluster: underrepresentation p < 10−14 in Up, enrichment p < 10−145 in Down. (C,D)—pluripotency signature (PluriNet) and regulation of multicellular organismal development GO:2000026 (MC development). PluriNet: enrichment p < 10−51 in Up, underrepresentation p < 10−11 in Down. MC development: underrepresentation p < 10−5 in Up; enrichment p < 10−28 in Down.
References
- Voskarides, K. Editorial: A New Bright Era for Evolutionary Medicine. J. Mol. Evol. 2020, 88, 1–2.
- Singh, P.P.; Isambert, H. OHNOLOGS v2: A Comprehensive Resource for the Genes Retained from Whole Genome Duplication in Vertebrates. Nucleic Acids Res. 2020, 48, D724–D730.
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “Virgin Birth”, Polyploidy, and the Origin of Cancer. Oncoscience 2014, 2, 3–14.
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered Interactions between Unicellular and Multicellular Genes Drive Hallmarks of Transformation in a Diverse Range of Solid Tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411.
- Niculescu, V.F. ACLS Cancers: Genomic and Epigenetic Changes Transform the Cell of Origin of Cancer into a Tumorigenic Pathogen of Unicellular Organization and Lifestyle. Gene 2020, 726, 144174.
- Fotiou, E.; Williams, S.; Martin-Geary, A.; Robertson, D.L.; Tenin, G.; Hentges, K.E.; Keavney, B. Integration of Large-Scale Genomic Data Sources With Evolutionary History Reveals Novel Genetic Loci for Congenital Heart Disease. Circ. Genom. Precis. Med. 2019, 12, 442–451.
- Arbabian, A.; Iftinca, M.; Altier, C.; Singh, P.P.; Isambert, H.; Coscoy, S. Mutations in Calmodulin-Binding Domains of TRPV4/6 Channels Confer Invasive Properties to Colon Adenocarcinoma Cells. Channels 2020, 14, 101–109.
- Yamasaki, M.; Makino, T.; Khor, S.-S.; Toyoda, H.; Miyagawa, T.; Liu, X.; Kuwabara, H.; Kano, Y.; Shimada, T.; Sugiyama, T.; et al. Sensitivity to Gene Dosage and Gene Expression Affects Genes with Copy Number Variants Observed among Neuropsychiatric Diseases. BMC Med. Genom. 2020, 13, 55.
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A Comprehensive Catalogue of Known and Candidate Cancer Genes from Cancer Sequencing Screens. Genome Biol. 2019, 20, 1.
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845–D855.
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary Framework of the Human Interactome: Unicellular and Multicellular Giant Clusters. Biosystems 2019, 181, 82–87.
- Dujon, A.M.; Aktipis, A.; Alix-Panabières, C.; Amend, S.R.; Boddy, A.M.; Brown, J.S.; Capp, J.-P.; DeGregori, J.; Ewald, P.; Gatenby, R.; et al. Identifying Key Questions in the Ecology and Evolution of Cancer. Evol. Appl. 2021, 14, 877–892.
- Pienta, K.J.; Hammarlund, E.U.; Brown, J.S.; Amend, S.R.; Axelrod, R.M. Cancer Recurrence and Lethality Are Enabled by Enhanced Survival and Reversible Cell Cycle Arrest of Polyaneuploid Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2020838118.
- Vinogradov, A.E. Human Transcriptome Nexuses: Basic-Eukaryotic and Metazoan. Genomics 2010, 95, 345–354.
- Vinogradov, A.E.; Anatskaya, O.V. Cell-Cycle Dependence of Transcriptome Gene Modules: Comparison of Regression Lines. FEBS J. 2020, 287, 4427–4439.
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. How the Evolution of Multicellularity Set the Stage for Cancer. Br. J. Cancer 2018, 118, 145–152.
- Liu, J. The Dualistic Origin of Human Tumors. Semin. Cancer Biol. 2018, 53, 1–16.
- Brock, A.; Huang, S. Precision Oncology: Between Vaguely Right and Precisely Wrong. Cancer Res. 2017, 77, 6473–6479.
- Shabo, I.; Svanvik, J.; Lindström, A.; Lechertier, T.; Trabulo, S.; Hulit, J.; Sparey, T.; Pawelek, J. Roles of Cell Fusion, Hybridization and Polyploid Cell Formation in Cancer Metastasis. World J. Clin. Oncol. 2020, 11, 121–135.
- Vinogradov, A.E.; Ezhevsky, S.A.; Rosanov, J.M.; Kazhdan, I.A.; Zweibach, A.S. Loosening of Cell Cycle Controls of Human Lymphocytes under the Action of Tumour Promoter TPA. Cell Prolif. 1991, 24, 493–505.
- Miles, D.M.; Desdouets, C.; Géli, V. Histone Stress: An Unexplored Source of Chromosomal Instability in Cancer? Curr. Genet. 2019, 65, 1081–1088.
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83.
- Dörnen, J.; Sieler, M.; Weiler, J.; Keil, S.; Dittmar, T. Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy. Int. J. Mol. Sci. 2020, 21, 1811.
- Cho, Y.; Kim, Y.K. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front. Oncol. 2020, 10, 764.
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14.
- Shu, Z.; Row, S.; Deng, W.-M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474.
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-Genome Doubling Confers Unique Genetic Vulnerabilities on Tumour Cells. Nature 2021, 590, 492–497.
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425.
More