Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Amina Yu and Version 3 by Amina Yu.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited disorder that typically presents in adults. Both kidneys are affected by cysts, leading to end stage renal disease in adulthood. Prevalence of ADPKD was estimated to be 1:400 to 1:1000 live births.
- autosomal dominant polycystic kidney disease
- cystogenesis
- therapy
1. Pathogenesis of Autosomal Dominant Polycystic Kidney Disease (ADPKD)
1.1. Polycystins and Signaling Pathways
The gene products of PKD1 and PKD2 genes are polycystin-1 (PC1) and polycystin-2 (PC2). PC1 and PC2 modulate number of signaling pathways in cooperation with many other proteins. Subcellularly, both proteins can be found in plasma membrane and PC2 in endoplasmic reticulum membrane. The assembly of PC1/PC2 complex obtained by cryo-electron microscopy was first presented in 2018 [1]. PC1/PC2 complex was found to be formed by one PC1 and three PC2 molecules. This observation suggests that due to three positively charged, cavity-facing residues of PC1 protein, cation permeation of the channel may be inhibited.
The typical manifestation of ADPKD is the formation of renal cysts. Cysts are formed in about 1% to 3% of nephrons [2]. Kidney cysts are formed already prenatally but they are mostly detected in adults [3]. The cyst development and enlargement include numerous cellular changes. The resulting changes in polycystin expression cause the damage of several intracellular signaling pathways terminating in the development of cysts due to cell proliferation and fluid secretion (Figure 1).
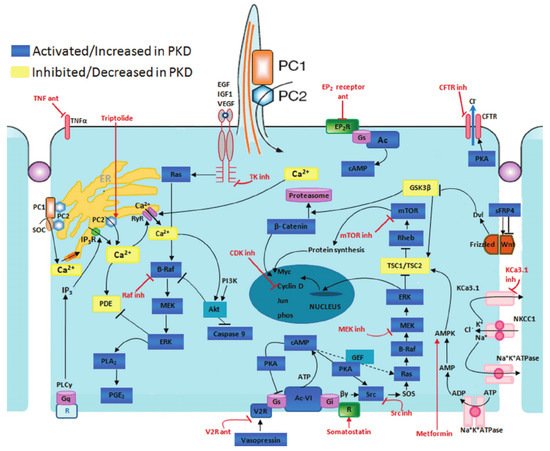
Figure 1. Disrupted pathways in polycystic kidney disease (PKD) and potential therapeutic intervention with specific agents. Agents associated with potential therapeutic interventions are represented in red; ant = antagonist; inh = inhibitor. AC-VI = adenylate cyclase 6; Akt = protein kinase B; AMPK = AMP kinase; B-Raf = v-raf murine sarcoma viral oncogene homolog B1; CDK = cyclin-de-pendent kinase; CFTR = cystic fibrosis transmembrane conductance regulator; EGF = epidermal growth factor; EP 2 R = E-prostanoid receptor 2; ERK = extracellular signal-regulated protein kinase; GSK3 = glycogen synthase kinase 3; IGF1 = insulin-like growth factor 1; IP 3 R = inositol 1,4,5-trisphosphate receptor; MAPK = mitogen-activated protein kinase; mTOR = mammalian target for rapamycin; PC1 = polycystin-1; PC2 = polycystin-2; PDE = phosphodiesterase; PGE 2 = prostaglandin E2; PI3K = phosphatidylinositol 3-kinase; PKA = protein kinase A; PLA 2 = phospholipase A2; PLCγ = phospholipase C-γ2; R = somatostatin sst2 receptor; Rheb = ras homolog enriched in brain; RyR = ryanodine receptor; sFRP4 = secreted Frizzled-related protein 4; TK = tyrosine kinase; TNF-α = tumor necro-sis factor-α; TSC = tuberous sclerosis proteins tuberin (TSC2) and hamartin (TSC1); V2R = vasopressin V2 receptor; VEGF = vascular endothelial growth factor [4].
1.2. Cyclic Adenosine Monophosphate (AMP) Pathway
One of the typical features of cyst-lining cells is elevated cAMP levels. cAMP stimulates the growth of cystic cells through stimulation of protein kinase A and activation of the Ras/Raf/ERK pathway leading to proliferation and enlargement of cystic cells. Cell proliferation of extracellular matrix is also increased. Moreover, cAMP increases the fluid secretion (involving cystic fibrosis transmembrane conductance regulator-CFTR) causing cyst growth [5]. Abundance of inflammatory cells and abnormal cell-cell junctions are typical for cystic tissue [6]. The most important therapeutical option is the reduction of cAMP in cysts which is associated with the decrease of the cyst growth.
1.3. The TSC-mTOR Pathway
The negative effect of PC1 on TSC-mTOR pathway (tuberous sclerosis complex-mammalian target of rapamycin) was described in [7]. TSC complex consisting of proteins TSC1 (also called hamartin) and TSC2 (also called tuberin) plays a role as a negative regulator of mTOR kinase, as it acts as a GTPase-activating protein. mTOR complex regulates cell growth and proliferation, as well as actin cytoskeleton and apoptosis.
Carboxy-terminal tail (CTT) of PC1 directly interacts with tuberin and protects it from phosphorylation by Akt (protein kinase B). PC1 leads to retention of TSC2 at plasma membrane which allows TSC2 to remain bound to TSC1 in functional complex [8]. On the other hand, TSC2 is essential for PC1 localization at plasma membrane [9].
1.4. PI3K/Akt Pathway
C-terminus of PC2 interacts with IP3 receptor (IP3R) on endoplasmic reticulum and can prolong IP3-dependent calcium release [10]. On the contrary, PC1 has an ability to inhibit this calcium release as it weakens the interaction between PC2 and IP3R [11]. It was shown that in the cell with PC1 expression the PI3K/Akt (Phosphatidylinositol-4,5-bisphosphate 3-kinase/Protein kinase B) pathway is activated. In this pathway, activated receptors located at plasma membrane directly stimulate PI3K triggering conversion of phosphatidylinositol-3,4-bis-phosphate (PIP2) to phosphatidylinositol-3,4,5-tris-phosphate (PIP3). Protein kinase B (Akt) binds to PIP3 at the plasma membrane which allows PDK1 (3-phosphoinositide dependent protein kinase 1) to activate Akt by its phosphorylation [12].
Activated Akt can influence apoptosis, growth, proliferation, metabolism, angiogenesis, survival, protein synthesis and transcription [13]. Activation of PI3K/Akt pathway may decrease in level of intracellular calcium. PC2 is subsequently recruited to the plasma membrane where it forms complex with PC1 serving as an influx calcium channel [13]. PC1 induces the resistance to apoptosis by activation PI3K [13].
1.5. The JAK-STAT Pathway
Polycystins play a role in the cell cycle. Both polycystins modulate JAK-STAT pathway (The Janus kinase-signal transducers and activators of transcription). PC1 binds and activates JAK2 kinase that subsequently phosphorylates STAT proteins (STAT1 and 3). Phosphorylated STAT1 shifts to the nucleus to bind to p21 gene promoter which is associated with higher gene expression. Increased levels of p21 prevent cells from proliferating by arrest in G0 phase of cell cycle. PC2 is required as a cofactor of this activation [14].
1.6. The Id Pathway
The Id pathway is associated with cell division. Id2 (Inhibitor of DNA binding 2) is a member of the helix-loop-helix family of proteins which bind to positively acting transcription factors and prevent them from binding to DNA. Putative binding partners for Id proteins were found to be transcription factors regulating cell such proteins as p21 [16]. PC2 can directly bind to Id2, Id2 is not able to bind E protein transcription. Moreover, the PC2-Id2 interaction is regulated by PC1-dependent serine phosphorylation of PC2 [17].
1.7. G-Protein Signaling Pathway
The cytosolic domain of PC1 activates transcription factor AP-1 (activating protein-1). AP-1 influences the cellular actions such as differentiation, proliferation and apoptosis [18]. Firstly, heterotrimeric G-proteins are activated by C-terminus of PC1. The activated Gα subunits activates c-Jun N-terminal kinase (JNK, member of MAPK kinases) that phosphorylates AP-1 transcription factor. In addition, JNK/AP-1 activation is supported by protein kinase C (PKC). AP-1 dependent transcription is triggered by PC2 interacting with PC1 [19].
Calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway regulates cell development and adaptation of different cell types [20]. PC1 activates trimeric G-protein that leads to Gα-dependent activation of phospholipase C (PLC) followed by release of calcium from endoplasmic reticulum. Increased Ca2+ concentration leads to activation of calcineurin, a Ca2+-dependent phosphatase. Dephosphorylated NFAT can enter into the nucleus and can influence the transcription of target genes [21].
Both families of AP-1 and NFAT transcription factors regulate to synergistically regulate gene expression of diverse genes with composite DNA elements containing adjacent NFAT and AP-1 binding sites [22]. This balanced activation is well described in the productive immune response but its possible effect on signaling pathways in other cells is yet to be proved.
1.8. Signaling Pathway
The Wnt signaling pathway regulates essential biological functions. Wnt proteins are growth factors. They play a roles in signaling pathways controlling proliferation, differentiation and cellular polarity during embryonal development [23]. It is divides into two major arms, the canonical Wnt/β-catenin pathway, and a β-catenin independent pathway that is responsible for establishing planar cell polarity and tissue morphogenesis.
In canonical Wnt pathway, secreted glycoproteins Wnt bind to Frizzled receptor (containing co-receptor LRP-low-density lipoprotein receptor-related protein). Activated Frizzled/LRP receptor can recruit cytoplasmatic protein Disheveled to the membrane providing a docking site for Axin and GSK-3 (glycogen synthase kinase 3) [24]. Axin targets β-catenin for ubiquitination. For activation of canonical Wnt pathway, β-catenin degradation is prevented. Therefore, the levels of β-catenin rise together with TCF (T cell factor) and promotes transcription of Wnt target genes [24]. Carboxy-terminal part of PC1 inhibits the interaction between β-catenin and TCF protein. The canonical Wnt pathway is activated in cystic tissue of ADPKD patients. PC1 could affect the cystogenesis during embryogenesis by the influence on Wnt pathway.
Noncanonical Wnt pathway plays role in determination of cellular polarity and motility and is required during tissue formation and homeostasis [25]. Activation of the noncanonical pathway leads to an increase of intracellular calcium. This pathway begins with the activation of the Frizzled receptor by Wnt ligands followed by G-protein-dependent activation of phospholipase C. Wnt can bind to the extracellular domain of PC1 which leads whole cell currents and Ca2+ influx dependent on PC2. Loss of cell polarity is frequently observed in cystic cells [26].
2. Treatments
2.1. Current Therapeutical Options
Healthy lifestyle and diet, maintenance of optimal weight, regular cardiovascular exercise and avoidance of smoking are generally recommended in ADPKD. High water intake is used as a prevention of nephrolithiasis but also leads to the suppression of vasopressin which could lead to an increase of fluid secretion into cysts. However, the results of trials with increased water intake were not conclusive. Salt intake should not exceed 5 g per day as all patients with other renal diseases.
Strict control of hypertension is the most important therapeutical option. Hypertension often presents in children and young adults with ADPKD and later promotes decline of renal function. Especially left ventricle hypertrophy was very common before greater attention was paid to treating blood pressure appropriately. RAS blockade is the mainstay of treatment of hypertension in ADPKD. The goal blood pressure should be less than 110/75 mm Hg in younger people than 50 years with eGFR above 60 mL/min per 1.73 m2. ACEI are recommended as the first safe choice [27]. The HALT one confirmed that the strict control of blood pressure decreases the mass of left ventricle, renal vascular resistance, proteinuria and kidney volume. Unfortunately, there was no significant effect of lower blood pressure on the slope of eGFR. The combination of ACEI and AT1 blockers was found to be safe even in ADPKD with advanced renal insufficiency but not more effective than monotherapy [28].
2.2. Tolvaptan
Tolvaptan is a selective arginine vasopressin type 2 receptor antagonist. Tolvaptan reduced the progression of kidney function decline in ADPKD in animal models [29][30]. The role of arginine vasopressin-mediated cAMP as a driver of fluid secretion into the cysts in ADPKD was demonstrated in preclinical ones. Two multicentric, randomized, placebo-controlled trials evaluated the safety and efficacy of tolvaptan in ADPDK patients. The TEMPO 3:4 trial (NCT00428948, ClinicalTrials.gov) enrolled 1445 ADPKD patients with normal renal function or mild renal insufficiency (CKD1, CKD, baseline eGFR ≥ 60 mL/min per 1.73 m2). Tolvaptan reduced an increase in kidney volume a 2.8% per compared with a 5.5% per year increase in the placebo group (p ˂ 0.001) over the 3-year period. Tolvaptan also slowed a decline in kidney function, as measured by reciprocal of serum creatinine level, compared with placebo, −2.61 vs. −3.81 per year (p ˂ 0.001) [31]. As expected, there were adverse aquaretic events, such as thirst, polyuria, nycturia, polakisuria and polydipsia. Moreover, clinically important liver involvement with increase of liver enzymes was described in 4.4% of patients. Liver enzymes in all patients normalized after cessation of tolvaptan. TEMPO 3:4 was followed by open label one TEMPO 4:4, the decrease of eGFR decline was found in the following two years [32]. A second trial, REPRISE (NCT02160145) was designed to evaluate the efficacy and safety of tolvaptan [33]. A one-year in ADPKD patients with more advanced disease (eGFR 25–65 mL/min per 1.73 m2) showed a reduction in decline of eGFR in treated patients. The post hoc analysis from open label extension found that tolvaptan also delayed eGFR decline in CKD4 (with eGFR 15 to 24 mL/min per 1.73 m2) [34]. A monthly protocol for monitoring liver enzymes is obligatory for 18 months as a prevention of severe hepatic toxicity, followed by 3-month check-up.
Nowadays, therapy with tolvaptan should be recommended to all ADPKD patients with probable rapid progression of the disease. Criteria for tolvaptan use are proposed as follows: 1. age 18–55 years, 2. chronic kidney disease CKD1–4 (eGFR ≥ 25 mL/min per 1.73 m2), 3. high risk measured by available risk scores (longitudinal diameter > 17 cm by ultrasound, total kidney volume > 750 mL, Mayo imaging classification 1C, 1D, 1E or PROPKD score > 6), and 4. rapid decline of eGFR 3 mL/min per > 1.73 m2) for 5 years. Mayo imaging classification is based on magnetic resonance and clearly predicts decline in eGFR on the basis of initial total kidney volume [35]. PROPKD (Predicting Renal Outcomes in Polycystic Kidney Disease) score predicts the prognosis according to molecular genetic analysis in combination with simple clinical information (sex, age at diagnosis of hypertension, age of first episode of macroscopic hematuria, flank pain related to cysts) [36]. Risk stratification is important because there was no difference of eGFR decline in patients having low risk of ADPKD progression [37]. Tolvaptan is not recommended in ADPKD patients with predicted mild clinical course.
2.3. Somatostatin Analogues
Somatostatin binds to five G protein-coupled receptors and inhibits directly and indirectly intracellular cAMP production in liver and kidney. This inhibition results in inhibition of fluid secretion, cell proliferation and induction of apoptosis. Somatostatin is rapidly eliminated and therefore analogues such as octreotide, lanreotide and pasireotide with a longer half-life have been synthesized. The efficacy of somastatin analogues on total kidney volume and on the decrease of eGFR was demonstrated in phase 2 small ones [38][39].
Octreotide did not significantly slow down the decline in eGFR and the increase of polycystic kidney volume after 3 years in a phase 3 trial ALADIN [40]. Somatostatin analogues led to decrease in GFR during first three months because of a decline in maladaptive hyperfiltration. Later, the effect is followed by a slower decline of eGFR because of the structural beneficial effect. Trials with such drugs require longer duration. A post hoc analysis of ALADIN confirmed that octreotide had a beneficial effect on the slope of measured GFR from the first to the third year of treatment.
A larger, open-label randomized controlled trial (DIPAK 1) was performed in the Netherlands, investigating the effects of lanreotide in 305 ADPKD patients with CKD3 after 2.5 years of treatment [41]. Lanreotide significantly reduced the growth of liver and kidney cysts. There was found no influence on eGFR. Nowadays, there are three with somatostatin ongoing or finalized.
On the other hand, promising data exists for somatostatin analogues for treatment of polycystic liver disease. In an open label, 6-month placebo-controlled trial of 27 patients treated with 120 mg of lanreotide versus 27 on placebo, lanreotide led to a 2.94% decrease of total liver volume compared with a 1.6% increase in placebo group [42]. In extension phase, the decrease of liver volume remained, but there was increase by 4% after cessation of treatment. Similarly, therapy with octreotide in an American one led to 4.7% decrease of liver volume [43]. Quality of life was also improved. Unfortunately, side-effects such as diarrhea, nausea, abdominal pain, meteorism, cholelithiasis and hyperglycemia can limit the use of somatostatin analogues. Higher prevalence of hepatic cyst infection was also reported.