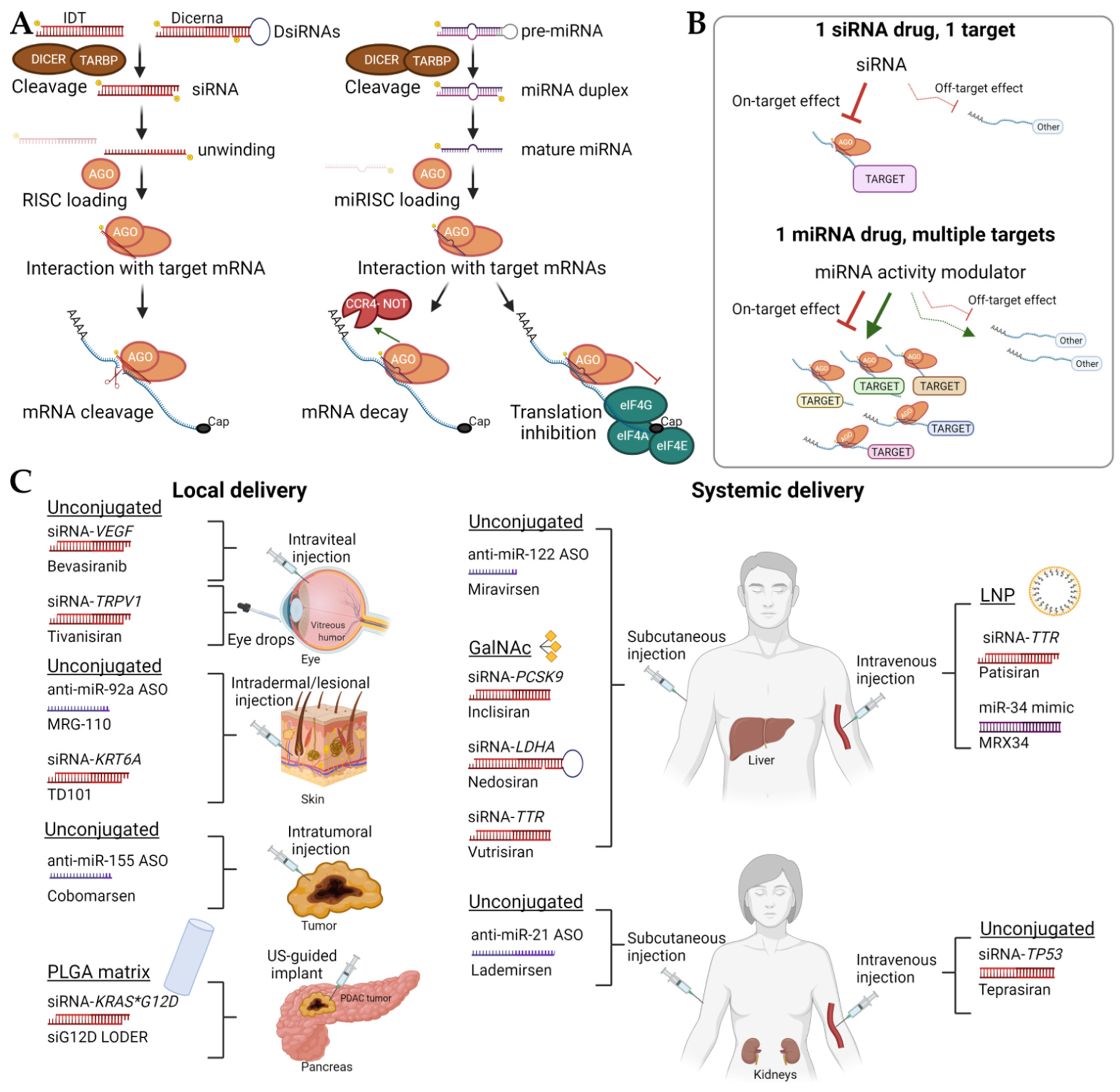
RNA therapies have demonstrated clinical potential for both the treatment of cancer and other pathologies. Therapeutic delivery and resulting adverse events remain significant roadblocks in implementing many of these drugs into clinical practice, but the FDA approval of three Alnylam Pharmaceuticals’ small interfering RNAs (siRNAs)RNA therapies has been a milestone in developing therapies tailored to disease-driving target genes. While it seems that RNAs can be administered “naked” in closed-compartment organs such as eyes and lungs, more research is needed for systemic administration. Lipid nanoparticles represent a promising delivery method, but some challenges remain because of their potential to elicit an immune response, relatively low circulation times, and relatively large size. The use of GalNAc for the delivery and targeting of siRNAs has made significant progress, but delivery systems targeted to organs other than the liver would broaden the range of diseases that could be treated with RNA therapies.
- RNA interferences
- microRNA
- siRNA
- drug delivery
- precision oncology
- nanomedicine
- nanoparticle
1. Unconjugated Short Non-Coding RNAs (ssncRNAs)
1.1. Diabetic Macular Edema and Age-Related Macular Degeneration
1.2. Respiratory Syncytial Infection
1.3. Pachyonychia Congenita
1.4. Hepatitis C
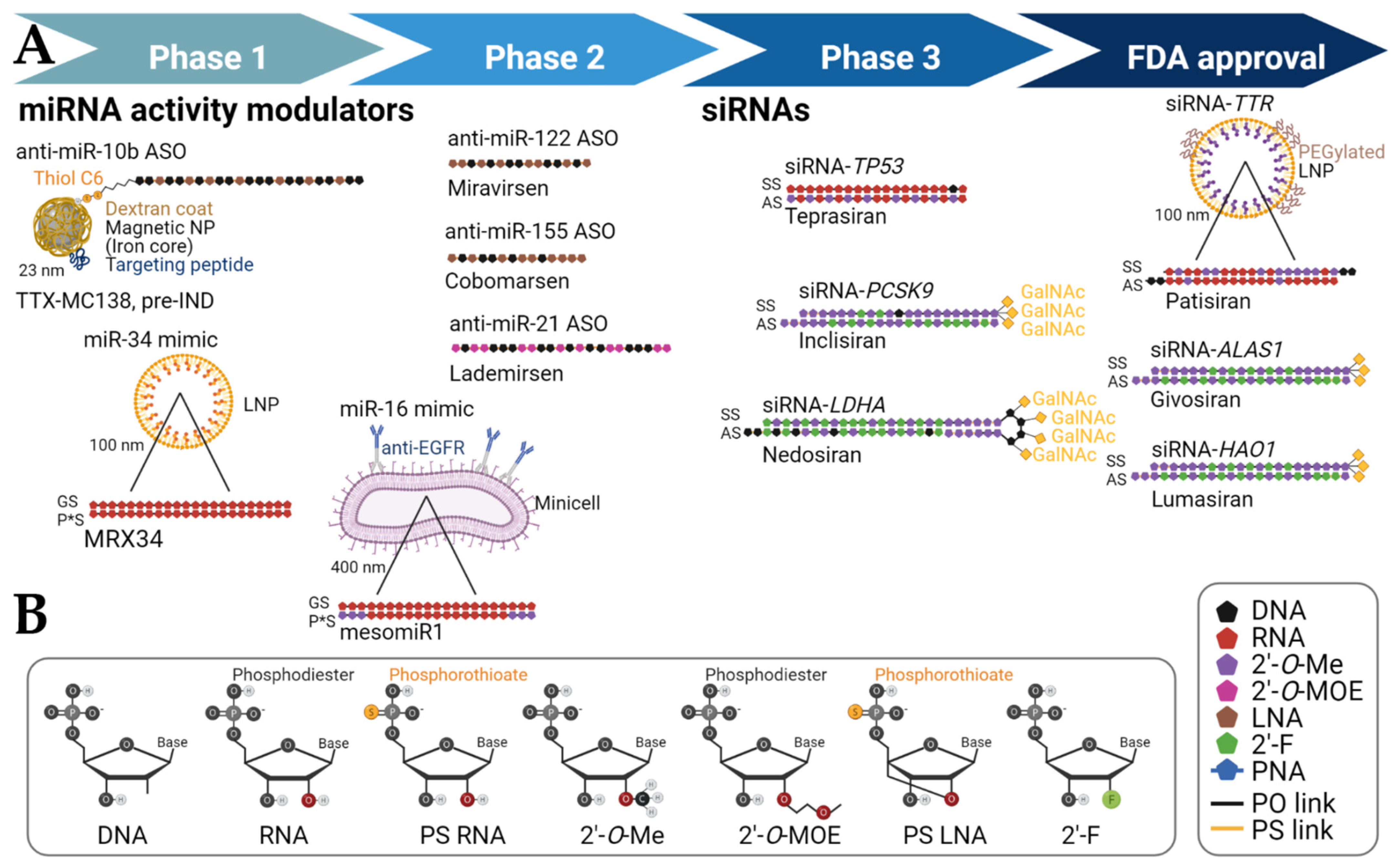
| miRNA Modulation |
Drug Name | Chemistry | Platform | Delivery | Disease (Organ Site) |
Sponsor | Clinical Status | References |
|---|---|---|---|---|---|---|---|---|
| miR-10b inhibition | RGLS5579 | ASO (2′-O-MOE, partial PS backbone) | - | Intravenous or intracranial | Glioblastoma (Brain) | Regulus Therapeutics (San Diego, CA, USA) | Pre-IND filing | [17] |
| miR-10b inhibition |
TTX-MC138 | ASO (partial LNA, partial PS backbone) |
Dextran-coated iron oxide magnetic nanoparticle | Intravenous | Metastatic breast cancer (Lung, other organs) | Transcode Therapeutics (Boston, MA, USA) | Pre-IND filing, scheduled 2022 | [18] |
| miR-16 restoration | mesomiR1 (TargomiR) |
dsRNA mimic (2′-O-Me on passenger strand only) | Bacterial minicells with anti-EGFR bispecific antibody | Intravenous | Recurrent malignant pleural mesothelioma and non-small cell lung cancer (Lung) | Asbestos Diseases Research Foundation (New South Wales, Australia), EnGeneIC Limited (Lane Cave West, Australia) | Phase 1 | NCT02369198, Competed |
| miR-21 inhibition | Lademirsen (SAR339375; previously known as RG-012 [Regulus]) | ASO (sugar 2′ position modifications, PS backbone) |
Unconjugated | Subcutaneous, 1.5 mg/kg | Alport syndrome (Kidney) | Genzyme, a Sanofi Company (Cambridge, MA, USA) | Phase 1 | NCT02855268, Completed |
| miR-21 inhibition | Lademirsen (SAR339375; previously known as RG-012 [Regulus]) | ASO (sugar 2′ position modifications, PS backbone) |
Unconjugated | Subcutaneous | Alport syndrome (Kidney) | Genzyme, a Sanofi Company (Cambridge, MA, USA) | Phase 2 | NCT02855268, Recruiting |
| miR-34a restoration | MRX34 | dsRNA mimic | Liposome | Intravenous | Primary liver cancer or other selected solid tumors or hematologic malignancies (Liver, other organs) | Mirna Therapeutics (Austin, TX, USA) | Phase 1 | NCT01829971, Terminated; NCT02862145, Withdrawn |
| miR-92a inhibition | MRG-110 | ASO (LNA-modified) | - | Intradermal | Wound healing | miRagen Therapeutics, Inc. (Boulder, CO, USA) | Phase 1 | NCT03603431, Completed |
| miR-122 inhibition | Miravirsen (SPC3649) | ASO (partial LNA, PS backbone) | Unconjugated | Subcutaneous | HCV chronic infection (Liver) | Copenhagen, Denmark | Phase 2 | NCT01200420, Completed |
| miR-155 inhibition | MRG-106 (Cobomarsen) | ASO (partial LNA) | Unconjugated | Intratumoral and/or intravenous or subcutaneous | Certain lymphomas and leukemias, including CTCL [mycosis fungoides subtype], CLL, DLBCL [activated B-cell (ABC) subtype], and ATLL | miRagen Therapeutics, Inc. (Boulder, CO, USA) | Phase 1 | NCT02580552, Completed |
| miR-155 inhibition | MRG-106 (Cobomarsen) | ASO (partial LNA) | Unconjugated | Intravenous | CTCL [mycosis fungoides subtype] | miRagen Therapeutics, Inc. (Boulder, CO, USA) | Phase 2 | NCT03713320 and NCT03837457, Terminated |
1.5. Acute Kidney Injury
1.6. Alport’s Disease
1.7. Cardiovascular Disease
1.8. Leukemias and Lymphomas
2. GalNAc-Conjugated sncRNAs
2.1. Porphyria
| Target Gene | Drug Name | Chemistry | Platform | Delivery | Treatment (Organ Site) |
Sponsor | References |
|---|---|---|---|---|---|---|---|
| ALAS1 | ALN-AS1 (Givosiran) * |
siRNA (2′-O-Me, 2′F, partial PS backbone) | GalNAc conjugation, 2.5 mg/kg | Subcutaneous | Acute Hepatic Porphyrias (Liver) |
Alnylam Pharmaceuticals (Cambridge, MA, USA) | NCT03338816, Completed |
| AT | Fitusiran ALN-AT3SC (Fitusiran) | siRNA (2′-O-Me, 2′-F, partial PS backbone) | GalNAc conjugation | Subcutaneous | Hemophilia A or B (Liver) | Genzyme, a Sanofi Company (Cambridge, MA, USA) | NCT03417102/03417245, Completed; NCT03754790/NCT03549871, Active |
| CASP2 | QPI-1007 | siRNA (2′-O-Me) | Up to 3 mg | Intraviteal | Acute Nonarteritic Anterior Ischemic Optic Neuropathy (Eye) | Quark Pharmaceuticals (Newark, CA, USA) | NCT02341560, Terminated |
| HAO1 | ALN-GO1 (Lumasiran) * | siRNA (2′-O-Me, 2′F, partial PS backbone) | GalNAc conjugation, up to 3 mg/kg | Subcutaneous | Primary Hyperoxaluria Type 1 (Liver) | Alnylam Pharmaceuticals (Cambridge, MA, USA) | NCT03681184, Active; NCT03905694, Active; NCT04152200, Active |
| LDHA | DCR-PHXC (Nedosiran) | DsiRNA pseudo-hairpin (2′-O-Me, 2′F, DNA, partial PS backbone) | GalXC | Subcutaneous | Hyperoxaluria (Liver) | Dicerna Pharmaceuticals (Lexington, MA, USA) | NCT04042402, Enrolling by invitation |
| PCSK9 | Inclisiran | siRNA (2′-O-Me, 2′F, internal DNA, partial PS backbone) | GalNAc conjugation, 300 mg | Subcutaneous | Homozygous Familial Hypercholesterolemia (Liver) | Novartis Pharmaceuticals (Basel, Switzerland) | NCT03851705, Active; NCT04659863, Recruiting |
| PCSK9 | Inclisiran | siRNA (2′-O-Me, 2′F, internal DNA, partial PS) | GalNAc conjugation, 300 mg | Subcutaneous | Atherosclerotic Cardiovascular Disease (ASCVD) or ASCVD High Risk and Elevated LDL-C (Liver) | Novartis Pharmaceuticals (Basel, Switzerland) | NCT04765657, Recruiting |
| PCSK9 | Inclisiran | siRNA (2′-O-Me, 2′F, internal DNA, partial PS backbone) | GalNAc conjugation, 300 mg | Subcutaneous | Prevent Cardiovascular events in Participants with Established Cardiovascular Disease (Liver) | Novartis Pharmaceuticals (Basel, Switzerland) | NCT05030428, Recruiting |
| TP53 | QPI-1002 (Teprasiran) | siRNA (2′-O-Me) | - | Intravenous | Improved Graft Function after Donor Kidney Transplant (Kidney) | Quark Pharmaceuticals (Newark, CA, USA) | NCT02610296, Completed |
| TP53 | QPI-1002 (Teprasiran) | siRNA (2′-O-Me) | - | Intravenous | Prevention of acute kidney injury after cardiac surgery (Kidney) | Quark Pharmaceuticals (Newark, CA, USA) | NCT03510897, Terminated |
| TRPV1 | SYL1001 (Tivanisiran) | siRNA | Ophthalmic solution | Periocular | Sjögren′s Syndrome, Dry eye (Eye) | Sylentis, S.A. (Madrid, Spain) | NCT04819269, Recruiting |
| TRPV1 | SYL1001 (Tivanisiran) | siRNA | Ophthalmic solution, 11.25 mg/mL | Periocular | Moderate to Severe Dry Eye Disease (Eye) | Sylentis, S.A. (Madrid, Spain) | NCT03108664, Completed |
| TTR | ALN-TTR02 (patisiran)* | siRNA (2′-O-Me, DNA overhangs) | Lipid nanoparticle | Intravenous | Transthyretin-Mediated Polyneuropathy (Liver) | Alnylam Pharmaceuticals (Cambridge, MA, USA) | NCT01960348, Completed |
| TTR | ALN-TTR02 (patisiran) | siRNA (2′-O-Me, DNA overhangs) | Lipid nanoparticle, 0.3 mg/kg | Intravenous | hATTR amyloidosis with disease progression after liver transplant (Liver) | Alnylam Pharmaceuticals (Cambridge, MA, USA) | NCT03862807, Completed |
| TTR | ALN-TTR02 (patisiran) | siRNA (2′-O-Me, DNA overhangs) | Lipid nanoparticle | Intravenous | ATTR Amyloidosis with Cardiomyopathy (Liver) | Alnylam Pharmaceuticals (Cambridge, MA, USA) | NCT03997383, Active |
| TTR | ALN-TTRSC (Revusiran) | siRNA (2′-O-Me, 2′-F) | GalNAc conjugation | Subcutaneous | Transthyretin-Mediated Familial Amyloidotic Cardiomyopathy (Liver) | Alnylam Pharmaceuticals (Cambridge, MA, USA) | NCT02319005, Completed |
| TTR | ALN-TTRSC02 (Vutrisiran) | siRNA (2′-O-Me, 2′-F, partial PS backbone) | GalNAc conjugation, 25 mg | Subcutaneous | Transthyretin Amyloidosis with Cardiomyopathy (Liver) | Alnylam Pharmaceuticals (Cambridge, MA, USA) | NCT04153149, Active |
| TTR | ALN-TTRSC02 (Vutrisiran) | siRNA (2′-O-Me, 2′-F, partial PS backbone) | GalNAc conjugation | Subcutaneous | hATTR Amyloidosis (Liver) | Alnylam Pharmaceuticals (Cambridge, MA, USA) | NCT03759379, Active |
| VEGF | Bevasiranib | siRNA | Up to 2.5 mg | Intraviteal | Age-Related Macular Degeneration following initiation of anti-VEGF Lucentis® antibody therapy (Eye) | OPKO Health, Inc. (Miami, FL, USA) | NCT00557791, Withdrawn |
2.2. Alpha-1 Antitrypsin Deficiency
2.3. Primary Hyperoxaluria
2.4. Hemophilia
2.5. Hepatitis B
2.6. Cholesterol Metabolism and Atherosclerotic Cardiovascular Disease
2.7. Atypical Hemolytic Uremic Syndrome
3. Lipid Nanoparticle Therapies
3.1. Transthyretin Amyloidosis
3.2. Liver Fibrosis
3.3. Hepatocarcinoma and Liver Metastases
3.4. Pancreatic Cancer