Several research disciplines require fast, reliable and highly automated determination of pharmaceutically active compounds and their enantiomers in complex biological matrices. To address some of the challenges of Capillary Electrophoresis (CE), such as low concentration sensitivity and performance degradation linked to the adsorption and interference of matrix components, CE in a hydrodynamically closed system was evaluated using the model compounds Pindolol and Propranolol. Some established validation parameters such as repeatability of injection, efficiency, resolution and sensitivity were used to assess its performance, and it was found to be broadly identical to that of hydrodynamically opened systems. While some reduction in separation efficiency was observed, this was mainly due to dispersion caused by injection and it had no impact on the ability to resolve enantiomers of model compounds even when spiked into complex biological matrix such as blood serum. An approximately 18- to 23-fold increase in concentration sensitivity due to the employment of wide bore capillaries was observed. This brings the sensitivity of CE to a level similar to that of liquid chromatography techniques. In addition to this benefit and unlike in hydrodynamically opened systems, suppression of electroosmotic flow, which is essential for hydrodynamically closed systems practically eliminates the matrix effects that are linked to protein adsorption.
1. Introduction
The determination of Active Pharmaceutical Ingredients (APIs) in biological fluids such as serum, plasma or urine is essential for several research disciplines such as drug pharmacology, drug metabolism, pharmacokinetics, as well as forensic and clinical toxicology. Routine, high throughput quantitative determinations of these compounds require that applied analytical techniques are sufficiently simple to perform while amenable to automation as well as providing adequate selectivity and sensitivity [1]. Capillary Electrophoresis (CE) is particularly suitable for the analysis of diverse types of APIs covering cationic, anionic as well as neutral analytes as it offers a wide range of separation modes in combination with a wide variety of detection techniques. Short analysis times, high resolution and high separation efficiency, in addition to low reagent costs and minimal sample requirements, make this technique particularly attractive [2][3][2,3]. Besides the obvious advantages, the analysis of APIs in complex biological samples can often be challenging due to multiple interferences from matrix components. An example of these are proteins, which are often present in these samples at very high concentrations and have a tendency to adsorb onto the inner wall of bare fused silica capillaries. This can lead to unpredictable fluctuations of electroosmotic flow (EOF) and consequently can cause highly variable migration velocities of analytes [4]. Slow rates of association and dissociation processes involved in the interaction of some of the analytes with proteins can significantly impair separation efficiencies due to electrodiffusion [5][6][5,6]. Additionally, biological fluids usually contain high concentrations of salts, which can have an adverse effect on the resolution of some analytes due to an increase in the electromigration dispersion of the matrix components. Besides high salt content, biological fluids also represent relatively complex systems with multiple chemical components present. This can lead to either an increased risk of comigrations with the analytes of interest especially when non-selective detection techniques are employed or signal suppression when mass selective detection is used. Due to low therapeutic concentrations of most pharmaceutical drugs their concentration levels in biological fluids are very low (ppb to ppm) [7]. Detection sensitivity of CE is, however, generally considered inferior to other separation techniques such as High-Performance Liquid Chromatography (HPLC). This is due to the restriction on sample injection volume (nL) because of the small internal diameter (i.d.) of the capillary tubes employed as separation compartments.
The vast majority of CE applications is carried out using capillaries with i.d. between 20–75 µm. This restriction is imposed due to the hydrodynamic flow dispersion [8], which becomes the prevalent cause of dispersion in capillaries of larger i.d. [9]. Additionally, larger i.d. capillaries are prone to excessive heat generation associated with the passage of electric current through them. This also contributes to zone dispersion, but in extreme cases, it can cause thermal degradation of analytes or breakdown of the current due to buffer boiling [10]. The moderate concentration sensitivity of current CE detection systems constitutes a significant challenge in achieving the required limits of detection (LOD) or quantitation for contemporary industrial applications. To overcome some of these challenges, extensive sample pre-treatment has become common practice in the analysis of biological fluids. This is carried out with two major objectives: (1) the removal of potentially interfering matrix components (sample clean-up); and (2) an increase of analyte concentration (sample pre- concentration).
An alternative way of increasing sample load-ability is to carry out CE analysis in wide bore (320 µm i.d.) capillary tubes. This wide bore capillary format also provides a longer optical path when compared to the conventionally used 50–75 µm i.d. capillaries thus improving detection sensitivity. To overcome the above-mentioned challenges posed by hydrodynamic dispersion, CE experiments employing wide bore capillaries can be performed in an hydrodynamically closed separation system (CSS). Such a system, originally developed for Capillary Isotachophoresis, but also applied in CE [8][11][12][8,11,12], prevents any hydrodynamically induced movement of the separation medium. In fact, under hydrodynamically closed conditions, any bulk movement of a separation medium must be prevented [11]. Papers dealing with various tools for effective and reproducible suppression of the EOF in CE capillaries can be found elsewhere [4][13][14][15][4,13,14,15].
2. Comparison of Hydrodynamically Opened and Closed Separation Systems
An hydrodynamically opened separation system (OSS) is the most common experimental setup for performing CE experiments in which reservoirs containing separation medium are exposed to atmospheric pressure. The capillary, which is filled with the separation medium (buffer), is fixed between these reservoirs. In this most basic experimental setup, analytes migrate from one end of the capillary (injection end) to the other end of the capillary (detection end) by a combination of electroosmotic movement and electrophoretic migration. The latter is facilitated by the analyte charge and electric field applied across the capillary. Separation of analyte zones is achieved by differences in their intrinsic electrophoretic mobilities (µep).
CE in OSS can be performed either with or without EOF. EOF is a flow of bulk liquid which originates from charge separation between the capillary wall and the separation medium. Its direction depends on the charge of the excess ions in the solution. If the electric field across the capillary is maintained constant by keeping the concentration of analytes low relative to the concentration of the separation buffer, then the EOF does not contribute to the dispersion of separated zones [8][16][8,16] due to its plug-like velocity profile. Under these circumstances EOF affects ion mobilities of all analytes equally, as it reduces or increases the magnitude of their electrophoretic velocity. Although EOF has a great influence on resolution in CE, many applications require its elimination in order to prevent electrostatic adsorption of analytes on capillary walls. EOF elimination can be achieved by means of either static chemical modification or dynamic capillary wall modification [4]. The latter approach is more flexible, and requires the addition of EOF suppressors to the separation medium [11][17][11,17]. Hydrodynamic flow (HDF) across a capillary, which can be generated by hydrostatic pressure difference between both capillary ends, is generally detrimental to the performance of CE as it adds to the dispersion of separated zones due to its parabolic velocity profile [18]. This flow is eliminated by ensuring that the level of fluid in both reservoirs is equal throughout the separation process and by limiting the i.d. of capillaries typically to 75 µm.
In hydrodynamically closed separation systems (CSS), the detection end of the capillary is usually separated from the buffer reservoir by a mechanical barrier in the form of a semipermeable membrane [18][19][18,19]. This means that one end of the capillary is practically blocked for the bulk of the separation medium as only ions can be electrophoretically transported across this membrane. This prevents the build-up of the hydrostatic pressure difference and eliminates HDF [18]. With CSS it is, therefore, possible to use capillaries with a larger i.d. As the zeta-potential of the membrane differs from that of the separation compartment, this gives rise to volume velocity differences and consequently increases the dispersion of separated zones through so called electroosmotic dispersion [18]. It is, therefore, necessary to suppress EOF when performing CE separations in CSS.
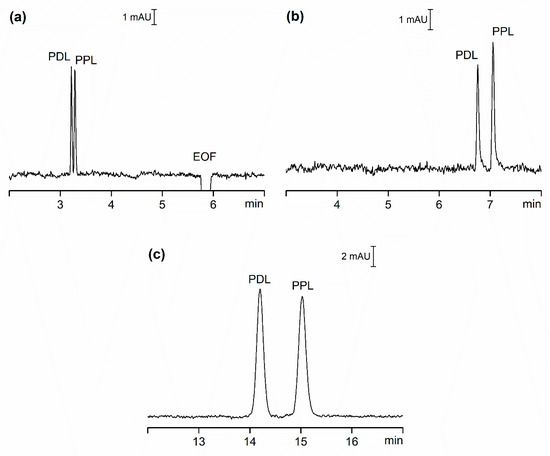
To compare OSS and CSS, we carried out CE analysis of two model pharmaceutical compounds, Pindolol (PDL) and Propranolol (PPL). Estimated basic pKa values of these compounds is approximately 9.5, calculated using ACD Percepta software (ACD/Labs, Toronto, ON, Canada), and at the pH of the separation buffer (pH = 3.7) they possess a positive charge. Figure 1 shows the separation of these two compounds under three scenarios: a) in OSS with EOF (OSS-EOF), b) in OSS with suppressed EOF (OSS-sEOF), and c) in CSS with suppressed EOF (CSS). The separation buffer for all systems was identical except for systems with suppressed EOF, where 0.05% (w/v) methylhydroxyethylcelullose (MHEC) was added. 0.1 mmol/L of diethylenetriamine (DETA) was also added to the separation buffer to prevent compound adsorption on the walls of separation compartments [20].
Figure 1. CE separation of PDL and PPL, each prepared at concentration of 5 µmol/L (CSS) and 20 µmol/L (OSS-EOF and OSS-sEOF). (a) OSS-EOF Buffer: 30 mmol/L β-alanine, 10 mmol/L poly(ethylene glycol) dicarboxylic acid, 0.1 mmol/L DETA; pH = 3.7; conditions: 25 kV (approximately 3.7 µA), 21 °C, detection at 210 nm. (b) OSS-sEOF Buffer: as (a) with addition of 0.05% MHEC; conditions: as (a). (c) CSS Buffer: as (b); conditions: as (a) except 75 µA (approximately 2.80 kV). See Materials and Methods for all other details.
In our attempt to compare the three systems, we focused on four criteria: repeatability of migration times, separation efficiency, resolution and sensitivity. To eliminate the impact of instrument set-up as much as possible we selected detectors with nearly identical setting for both OSS and CSS experiments. Table 1 lists some of the data for these three systems expressed as the average of ten repeated injections of the model sample.
Table 1. Some analytical performance data for the separations of PDL and PPL in the opened (OSS-EOF and OSS-sEOF) and closed (CSS) separation systems.
| Parameter |
OSS-EOF |
OSS-sEOF |
CSS |
| |
PDL |
PPL |
PDL |
PPL |
PDL |
PPL |
| RSD tm (%) |
0.3 |
0.3 |
0.4 |
0.4 |
0.5 |
0.5 |
| RSD A (%) |
2.0 |
1.4 |
3.4 |
2.4 |
2.2 |
1.6 |
| N × 10 | −3 | (m | −1 | ) |
309 |
310 |
465 |
482 |
162 |
147 |
| LOD (µmol/L) |
1.20 |
1.10 |
1.50 |
1.40 |
0.06 |
0.07 |
| Resolution | (1) |
1.47 |
3.64 |
3.23 |
References
- Yıldırım, S.; Erkmen, C.; Uslu, B. Novel Trends in Analytical Methods for β-Blockers: An Overview of Applications in the Last Decade. Crit. Rev. Anal. Chem. 2020, 1–39.
- El Deeb, S.; Wätzig, H.; El-Hady, D.A.; Van De Griend, C.E.S.; Scriba, G.K.E. Recent advances in capillary electrophoretic migration techniques for pharmaceutical analysis (2013–2015). Electrophoresis 2016, 37, 1591–1608.
- Phillips, T.M. Recent advances in CE and microchip-CE in clinical applications: 2014 to mid-2017. Electrophoresis 2018, 39, 126–135.
- Huhn, C.; Ramautar, R.; Wuhrer, M.; Somsen, G.W. Relevance and use of capillary coatings in capillary electrophoresis-mass spectrometry. Anal. Bioanal. Chem. 2010, 396, 297–314.
- Gaš, B.; Kenndler, E. Dispersive phenomena in electromigration separation methods. Electrophoresis 2000, 21, 3888–3897.
- Gaš, B.; Štědrý, M.; Kenndler, E. Peak broadening in capillary zone electrophoresis. Electrophoresis 1997, 18, 2123–2133.
- Junaid, T.; Wu, X.; Thanukrishnan, H.; Venkataramanan, R. Therapeutic Drug Monitoring. In Clinical Pharmacy Education, Practice and Research; Thomas, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 425–436.
- Virtanen, R. Zone electrophoresis in narrow-bore tube employing potentiometric detection. A theoretical and experimental study. Acta Polytech. Scand. Chem. Technol. Ser. 1974, 123, 1–67.
- Yin, H.; Keely-Templin, C.; McManigill, D. Preparative capillary electrophoresis with wide-bore capillaries. J. Chromatogr. A 1996, 744, 45–54.
- Valkó, I.E.; Porras, S.P.; Riekkola, M.-L. Capillary electrophoresis with wide-bore capillaries and non-aqueous media. J. Chromatogr. A 1998, 813, 179–186.
- Kaniansky, D.; Masár, M.; Bielčı́ková, J. Electroosmotic flow suppressing additives for capillary zone electrophoresis in a hydrodynamically closed separation system. J. Chromatogr. A 1997, 792, 483–494.
- Mikkers, F.; Everaerts, F.; Verheggen, T. Concentration distributions in free zone electrophoresis. J. Chromatogr. A 1979, 169, 1–10.
- Ramos-Payán, M.; Ocaña-Gonzalez, J.A.; Fernández-Torres, R.M.; Llobera, A.; Bello-López, M.Á. Recent trends in capillary electrophoresis for complex samples analysis: A review. Electrophoresis 2018, 39, 111–125.
- Hajba, L.; Guttman, A. Recent advances in column coatings for capillary electrophoresis of proteins. TrAC Trends Anal. Chem. 2017, 90, 38–44.
- Štěpánová, S.; Kašička, V. Recent applications of capillary electromigration methods to separation and analysis of proteins. Anal. Chim. Acta 2016, 933, 23–42.
- Reijenga, J.; Kenndler, E. Computational simulation of migration and dispersion in free capillary zone electrophoresis. J. Chromatogr. A 1994, 659, 403–415.
- Wang, A.-J.; Feng, J.-J.; Dong, W.-J.; Lu, Y.-H.; Li, Z.-H.; Riekkola, M.-L. Spermine-graft-dextran non-covalent copolymer as coating material in separation of basic proteins and neurotransmitters by capillary electrophoresis. J. Chromatogr. A 2010, 1217, 5130–5136.
- Kaniansky, D.; Marak, J.; Masár, M.; Iványi, F.; Madajová, V.; Šimuničová, E.; Zelenská, V. Capillary zone electrophoresis in a hydrodynamically closed separation system with enhanced sample loadability. J. Chromatogr. A 1997, 772, 103–114.
- Foret, F.; Křivánková, L.; Boček, P. Capillary Zone Electrophoresis; VCH: Weinheim, Germany, 1993.
- Lucy, C.A.; Macdonald, A.M.; Gulcev, M.D. Non-covalent capillary coatings for protein separations in capillary electrophoresis. J. Chromatogr. A 2008, 1184, 81–105.