KMT2A (Lysine methyltransferase 2A) is a member of the epigenetic machinery, encoding a lysine methyltransferase responsible for the transcriptional activation through lysine 4 of histone 3 (H3K4) methylation. KMT2A has a crucial role in gene expression, thus it is associated to pathological conditions when found mutated. KMT2A germinal mutations are associated to Wiedemann–Steiner syndrome and also in patients with initial clinical diagnosis of several other chromatinopathies (i.e., Coffin–Siris syndromes, Kabuki syndrome, Cornelia De Lange syndrome, Rubinstein–Taybi syndrome), sharing an overlapping phenotype. On the other hand, KMT2A somatic mutations have been reported in several tumors, mainly blood malignancies. Due to its evolutionary conservation, the role of KMT2A in embryonic development, hematopoiesis and neurodevelopment has been explored in different animal models, and epigenetic treatments for disorders linked to KMT2A dysfunction have been extensively investigated.
1. Introduction
KMT2A (Lysine methyltransferase 2A), also known as MLL1, is a protein coding gene mapping to human chromosome 11 (11q23.3), made up of 90,343 bases (GRCh38/hg38) and 37 exons belonging to KMTs (Lysine methyltransferases) family.
KMTs catalyze the transfer of methyl groups from S-adenosylmethionine to the lysine residues on histone tails, particularly the histone H3 tail. Unlike other epigenetic enzymes such as acetyltransferases (HATs), KMTs are more specific and usually modify one or two lysines on a single histone
[1]. The effect on chromatin state, i.e., whether it activates transcription or represses it, depends on the methylation states and their positions (
Figure 1)
[2][3][4][5][6][7][8][9][10][11][12][13][14][15]. KMTs are so called writers, enzymes that catalyze the addition of chemical groups to histone tails or to DNA; these modifications are not permanent but can be removed by erasers to reverse the influence on gene expression. Readers possess specialized domains able to recognize and interpret different chemical modifications. Writers, erasers and readers form the epigenetic machinery, and mutations in genes coding for this apparatus lead to ann altered chromatin conformation and an incorrect gene expression, resulting in a series of syndromes known as chromatinopathies, Mendelian genetic diseases, most of them with a dominant character
[16][17][18].
Pathogenic mutations in KMTs and KDMs (Lysine demethylases) lead to haploinsufficiency in numerous developmental syndromes (
Figure 2) (
Table 1)
[10][19].
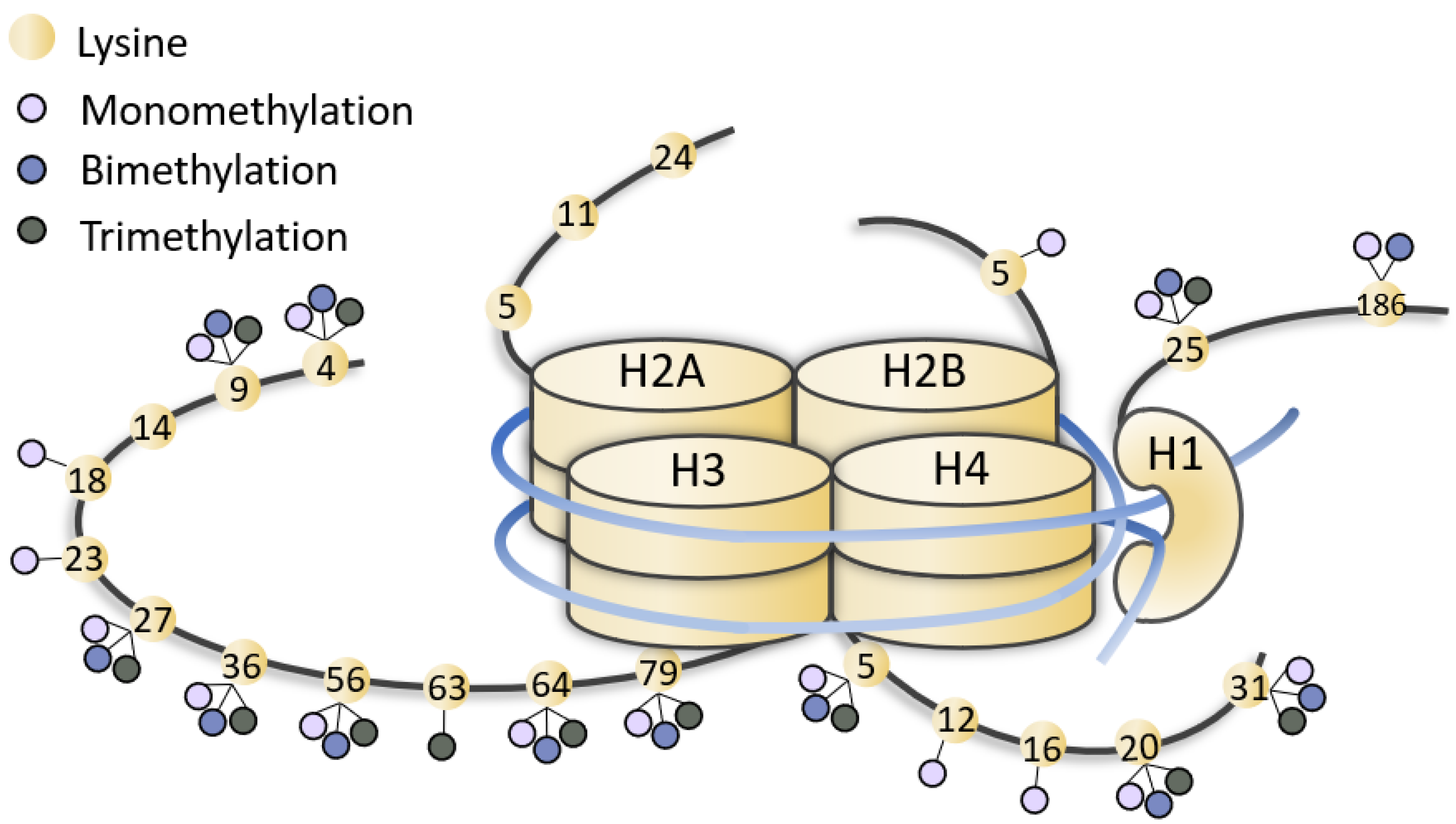
Figure 1. Representation of methylated lysines of histone tails. Lysines (yellow dots) of histone (H1, H2, H3, H4) tails can be mono-, bi-, tri-methylated (little lilac, blue and grey dots). The figure is not drawn to scale.
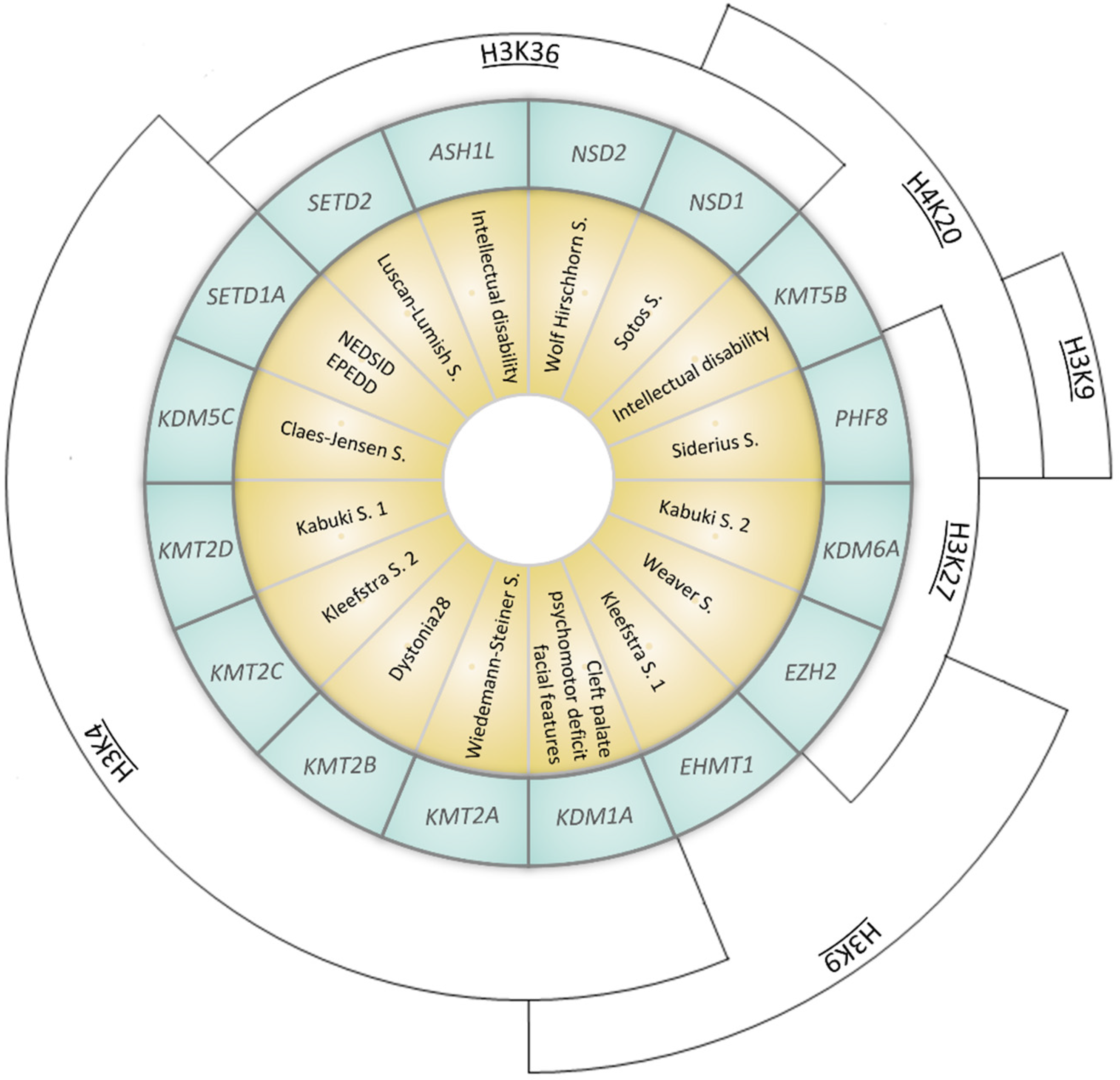
Figure 2. Representation of syndromes caused by mutations in genes coding for KMTs (Lysine methyltransferases) or KDMs (Lysisne demethylases). Syndromes (yellow inner ring) and the corresponding causative gene (coding for KMTs or KDMs, listed in the middle blue ring) are represented. The outer arcs indicate the site of epigenetic modification (NEDSID: Neurodevelopmental disorder with speech impairment and dysmorphic facies; EPEDD: Epilepsy, early-onset, with or without developmental delay).
Table 1. Details of genes and syndromes represented in Figure 2.
| Gene (OMIM *) |
Associated Developmental Disorder(s) (OMIM #) |
Targeted Lysine Residue |
| SETD1A (611052) |
Neurodevelopmental disorder with speech impairment and dysmorphic facies NEDSID (619056)/Epilepsy, early-onset, with or without developmental delay EPEDD (618832) |
H3K4 (met) |
| SETD2 (612778) |
Luscan-Lumish S. (616831) |
H3K36 (met) |
| KDM1A (609132) |
Cleft palate, psychomotor retardation, distinctive facial features (616728) |
H3K4 (demet)
H3K9 (demet) |
| KDM5C (314690) |
Claes-Jensen S. (300534) |
H3K4 (demet) |
[22].
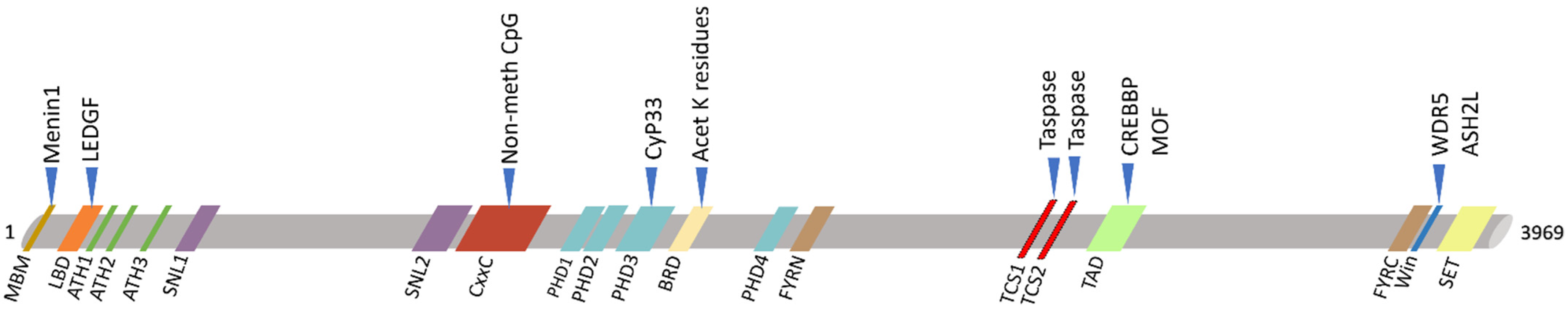
Figure 3. Schematic view of KMT2A protein domains (below) and its main interactors (upper). KMT2A domains: MBM, high-affinity Menin-binding motif, residues 6–10; LBD, LEDGF-binding domain, residues 109–153; ATH1-2-3, AT-Hook1/2/3, residue 169–180, residues 217–227, residue 301–309; SNL1-2, nuclear-localization signal 1/2, residues 400–443, residues 1008–1106; CxxC, including: pre-CxxC region, residues 1149–1154, CxxC domain, residues 1147–1242, post-CxxC residues 1298–1337; PHD1-2-3-4, plant homology domain 1/2/3/4, residues 1431–1482, residues 1479–1533, residues 1566–1627, residues 1931–1978; BRD, bromodomain, residues 1703–1748; FYRN, FY-rich N-terminal domain, residues 2018–2074; TAD, transactivator domain, residues 2829–2883; FYRC, FY-rich C-terminal domain, residues 3666–3747; Win, WDR5 interaction motif, residues 3762–3773; SET, Su(Var)3-9 enhancer-of-zeste trithorax domain, residues 3829–2945. KMT2A has two sites for cutting by Taspase1: TCS1-2, taspase1 cleavage site 1/2, residue 2666–2670 and residues 2718–2722.
As other members of KMTs family, KMT2A regulates gene transcription through chromatin opening or closure and its activity is antagonized by the lysine demethylases (KDMs) family.
2. KMT2A Germline Mutations
2.1. Wiedemann–Steiner Syndrome
KMT2A germinal variants are associated to the Wiedemann–Steiner syndrome (WDSTS, OMIM #605130), a rare autosomal dominant disorder characterized by different features, mainly intellectual disability (ID), developmental delay (DD), pre- and post-natal growth deficiency, hypertrichosis, short stature, hypotonia, distinctive facial features (thick eyebrows, long eyelashes, narrow palpebral fissures, broad nasal tip, down slanting palpebral fissures), skeletal abnormalities (clinodactyly, brachydactyly, accelerated skeletal maturation), feeding problems and behavioral difficulties (
Table 2)
[23][24][25].
KMT2A variants are distributed throughout the gene, with a pathogenic mutation hotspot in exon 27, and most of them lead to
KMT2A loss of function. WDSTS patients usually present de novo private mutations, and the diagnosis is based on clinical evaluation of signs and symptoms then confirmed by molecular analysis. Unfortunately, a specific treatment is not available, thus possible interventions aim at reducing the severity of symptoms.
Table 2. Clinical signs reported in patients with a KMT2A mutation and an initial clinical diagnosis of chromatinopathy. Presence of all features is compared with the one in WDSTS.
| |
WDSTS |
CdLS |
CSS |
KS |
RSTS |
| [26] |
1 + 1 pt [27][28] |
1 pt [29] |
2 pt [30] |
1 + 6 pt [26][31] |
| Vision problems |
− |
0/2 |
1/1 |
1/2 |
1/7 |
| Cardiac problems |
+ |
1/2 |
1/1 |
1/2 |
0/7 |
| CNS problems |
+/− |
1/2 |
0/1 |
NA |
0/7 |
| Genitourinary problems |
− |
0/2 |
1/1 |
1/2 |
2/7 |
KDM6A (300128) |
Kabuki S. 2 (300867) |
H3K27 (demet) |
| Feeding problems |
+ |
0/2 |
1/1 |
1/2 |
3/7 |
KMT2A (159555) |
Wiedemann–Steiner S. (605130) |
| Behavior problems | H3K4 (met) |
| + |
1/2 |
0/1 |
NA |
3/7 |
KMT2B (606834) |
Dystonia 28 (617284) |
H3K4 (met) |
| Frequent infection |
− |
0/2 |
1/1 |
1/2 |
0/7 |
KMT2C (606833) |
Kleefstra S. 2 (617768) |
H3K4 (met) |
| Seizures |
+/− |
0/2 |
0/1 |
1/2 |
1/7 |
KMT2D (602113) |
Kabuki S. 1 (147920) |
H3K4 (met) |
| ID |
++ |
2/2 |
1/1 |
1/2 |
KMT5B (610881) |
Intellectual disability (617788) |
H4K20 (met) |
| EZH2 (601573) |
Weaver S. (277590) |
H3K9 (met)
H3K27 (met) |
| EHMT1 (607001) |
Kleefstra S. 1 (610253) |
H3K9 (met) |
| ASH1L (607999) |
Intellectual disability (617796) |
H3K36 (met) |
| NSD1 (606681) |
Sotos S. (117550) |
H3K36 (met)
H4K20 (met) |
| NSD2 (602952) |
Wolf Hirschhorn S. (194190) |
H3K36 (met) |
| PHF8 (300560) |
Siderius S. (300263) |
H3K9 (demet)
H3K27 (demet)
H4K20 (demet) |
*: Gene, #: Associated Developmental Disorder(s).
Many species have a
KMT2A ortholog, including fishes, birds, amphibians, and mammals; thus, its evolutionary conservation allowed a comprehensive study of KMT2A molecular functions through in vivo experiments on animal models (
Drosophila melanogaster, Danio rerio, Mus musculus).
KMT2A expression is mainly nuclear and ubiquitously present in 27 tissues, especially in ovary, lymph node, endometrium, thyroid and brain tissue
[20].
KMT2A encodes a lysine methyltransferase (KMT) formed of 3969 amino acids, a transcriptional co-activator which plays a crucial role in hematopoiesis, in regulating gene expression at early developmental stages, and in the control of circadian gene expression. KMT2A is processed by the endopeptidase Taspase 1 in two fragments (MLL-C and MLL-N) which heterodimerize and regulate the transcription of specific genes, including
HOX genes
[21]. KMT2A protein has 18 domains, including the CXXC-type zinc finger, the extended PHD domain and the bromodomain. The SET domain has the methyltransferase activity (mono-, di-, tri-methylation) on lysine 4 of histone 3 (H3K4 me1/2/3), a post-transcriptional modification (PTM) responsible of epigenetic transcriptional activation and which efficiency can be increased when the protein is associated with another component of the MLL1/MLL complex (
Figure 3)
| 7/7 |
| Speech delay |
| ++ |
| 1/2 |
| 1/1 |
NA |
5/7 |
| Microcephaly |
− |
2/2 |
NA |
NA |
3/7 |
| Eyes anomalies (thick eyebrows, synophrys, long eyelashes, ptosis, downslanting/narrow palpebral fissure) |
+ |
2/2 |
1/1 |
2/2 |
7/7 |
| Nose anomalies (depressed nasal bridge, broad nasal tip) |
+ |
2/2 |
1/1 |
2/2 |
7/7 |
| Mouth anomalies (high arched palate, thin upper vermilion) |
+/− |
2/2 |
1/1 |
0/2 |
4/7 |
| Hands/feet anomalies (clinodactyly, brachydactyly, persistent fetal finger pads, broad halluces) |
+/− |
2/2 |
1/1 |
2/2 |
6/7 |
| Delayed bone age |
+ |
0/2 |
NA |
NA |
0/7 |
| Hirsutism |
+ |
1/2 |
1/1 |
NA |
4/7 |
| Hypotonia |
++ |
NA |
1/1 |
2/2 |
3/7 |
WDSTS: Wiedemann–Steiner syndrome, CdLS: Cornelia De Lange syndrome, CSS: Coffin–Siris syndromes, KS: Kabuki syndrome, RSTS: Rubinstein–Taybi syndrome, CNS: Central nervous system, ID: intellectual disability. ++ = 70–100% WDSTS patients; + = 20–70% WDSTS patients; +/− = 5–20% WDSTS patients; − = <5% WDSTS patients; NA = not assessed.
2.2. Other Chromatinopathies
Mutations in KMT2A have been also found in patients with a clinical presentation suggestive of other chromatinopathies (i.e., Coffin–Siris syndromes, Kabuki syndrome, Cornelia De Lange syndrome, Rubinstein–Taybi syndrome) but negative for alterations in the related known-causative genes. Their clinical presentation shares with WDSTS some phenotypic features and it is caused by alterations of genes involved in the regulation and maintenance of chromatin state as KMT2A. Indeed, these syndromes are caused by mutations in genes of the epigenetic machinery and therefore are known as chromatinopathies [16][18][16,18].
Table 2. Clinical signs reported in patients with a KMT2A mutation and an initial clinical diagnosis of chromatinopathy. Presence of all features is compared with the one in WDSTS.
| |
WDSTS |
CdLS |
CSS |
KS |
RSTS |
| [26] |
1 + 1 pt [27][28] |
1 pt [29] |
2 pt [30] |
1 + 6 pt [26][31] |
| Vision problems |
− |
0/2 |
1/1 |
1/2 |
1/7 |
| Cardiac problems |
+ |
1/2 |
1/1 |
1/2 |
0/7 |
| CNS problems |
+/− |
1/2 |
0/1 |
NA |
0/7 |
| Genitourinary problems |
− |
0/2 |
1/1 |
1/2 |
2/7 |
| Feeding problems |
+ |
0/2 |
1/1 |
1/2 |
3/7 |
| Behavior problems |
+ |
1/2 |
0/1 |
NA |
3/7 |
| Frequent infection |
− |
0/2 |
1/1 |
1/2 |
0/7 |
| Seizures |
+/− |
0/2 |
0/1 |
1/2 |
1/7 |
| ID |
++ |
2/2 |
1/1 |
1/2 |
7/7 |
| Speech delay |
++ |
1/2 |
1/1 |
NA |
5/7 |
| Microcephaly |
− |
2/2 |
NA |
NA |
3/7 |
| Eyes anomalies (thick eyebrows, synophrys, long eyelashes, ptosis, downslanting/narrow palpebral fissure) |
+ |
2/2 |
1/1 |
2/2 |
7/7 |
| Nose anomalies (depressed nasal bridge, broad nasal tip) |
+ |
2/2 |
1/1 |
2/2 |
7/7 |
| Mouth anomalies (high arched palate, thin upper vermilion) |
+/− |
2/2 |
1/1 |
0/2 |
4/7 |
| Hands/feet anomalies (clinodactyly, brachydactyly, persistent fetal finger pads, broad halluces) |
+/− |
2/2 |
1/1 |
2/2 |
6/7 |
| Delayed bone age |
+ |
0/2 |
NA |
NA |
0/7 |
| Hirsutism |
+ |
1/2 |
1/1 |
NA |
4/7 |
| Hypotonia |
++ |
NA |
1/1 |
2/2 |
3/7 |
WDSTS: Wiedemann–Steiner syndrome, CdLS: Cornelia De Lange syndrome, CSS: Coffin–Siris syndromes, KS: Kabuki syndrome, RSTS: Rubinstein–Taybi syndrome, CNS: Central nervous system, ID: intellectual disability. ++ = 70–100% WDSTS patients; + = 20–70% WDSTS patients; +/− = 5–20% WDSTS patients; − = <5% WDSTS patients; NA = not assessed.
3. KMT2A Somatic Mutations
KMT2A somatic mutations are implicated in several tumors. The most common types of alterations involving
KMT2A are mutations (3.62%), fusions (0.13%) (with more than 80 different partners identified)
[32], losses (0.10%), amplifications (0.07%), and
KMT2A-EP300 fusions (0.19%)
[33]. Among the mutations, the most frequent observed in patient-derived samples are missense (54.36%), synonymous (13.61%) and nonsense substitutions (7.34%)
[34]. On the contrary, in patients with germline
KMT2A mutations, the ones more represented are frameshift (41%) and stop mutations (29%), followed by missense variants (18%)
[26]. The project GENIE, led by the American Association for Cancer Research, highlighted how
KMT2A is implicated in many diseases, especially in blood cancers such as acute myeloid leukemia (2.49%), T-cell lymphoblastic leukemia (5.63%) and up to 14% in high grade B-cell lymphoma (
Figure 5).
KMT2A is altered also in 4.65% of malignant solid tumors, such as lung adenocarcinoma, colon adenocarcinoma and bladder urothelial carcinoma (
Figure 5). Interestingly,
KMT2A is not the only gene of the epigenetic apparatus whose somatic mutations give rise to tumors, especially in blood malignancies. In fact, somatic alterations in other chromatinopathies genes were found in myelodysplastic syndromes (
ASXL1, ATRX, DNMT3A, EED, EZH2, KDM6A, KMT2 family genes,
PHF6)
[35], acute myeloid leukemia (
ASXL1, DNMT3A, PHF6)
[36], multiple myeloma (
KDM6A, KMT2B, KMT2C, WHSC1)
[37] and lymphoid malignancies such as acute lymphoblastic leukemia (
CREBBP, DNMT3A, EP300, EED, EZH2, PHF6) and diffuse large B-cell lymphoma (
CREBBP/EP300, EZH2, KMT2C/D)
[37][38].
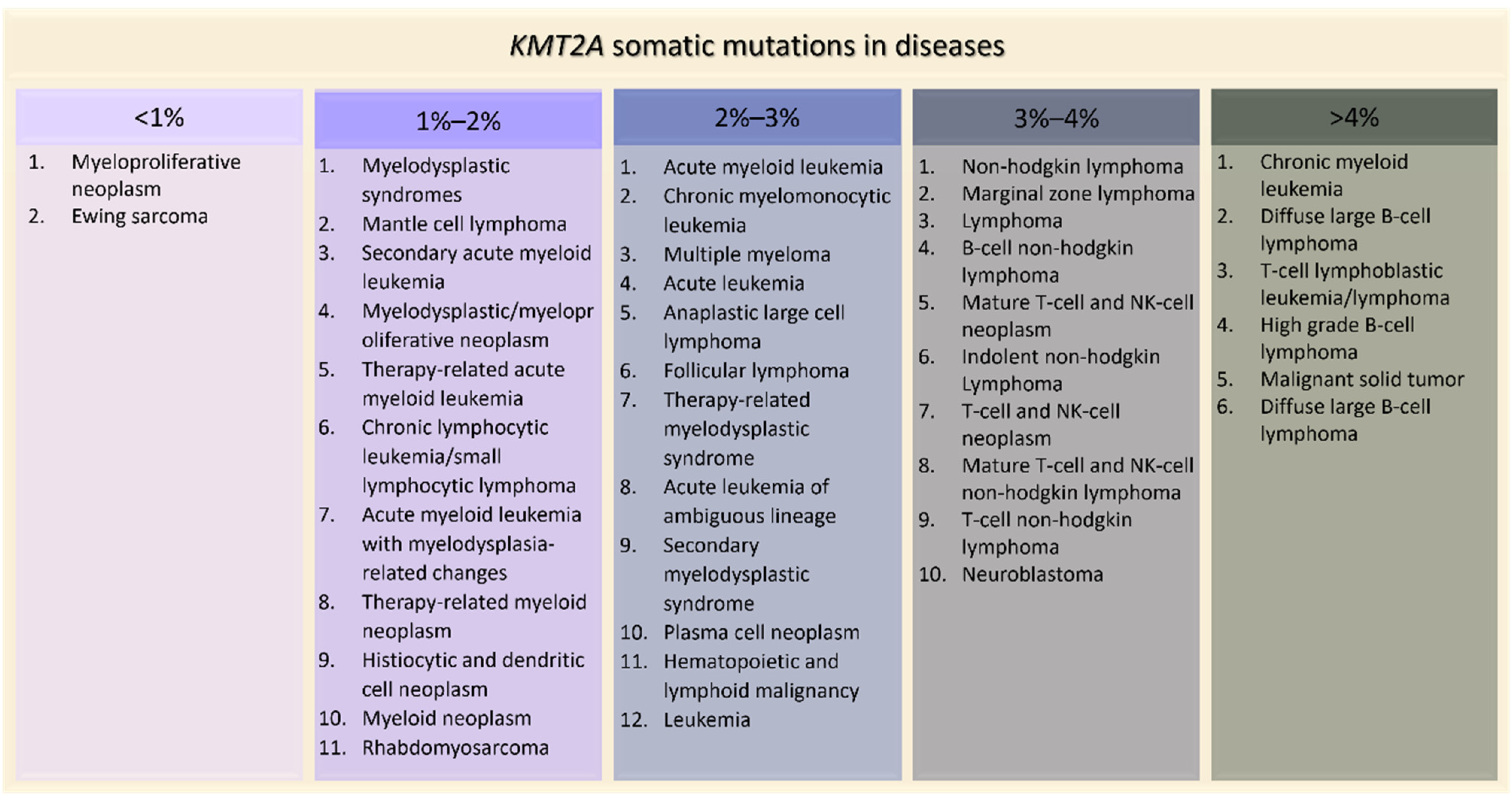 Figure 5. KMT2A
Figure 5. KMT2A somatic mutations in tumors ordered by percentage of positive cases (AACR Project GENIE).
4. Effects of KMT2A Mutations in Animal Models
KMT2A is an evolutionary conserved gene, involved in several functional process of embryonic development, ranging from hematopoiesis to neurogenesis. Indeed, in 1995, Yu and colleagues showed that the complete disruption of
KMT2A was embryonic lethal in mice, and heterozygous animals were anemic and affected by growth delay, hematopoietic anomalies and skeletal malformations
[39]. Developmental defects were investigated in
Drosophila melanogaster too, where mutations in
KMT2A homolog (
trx) led to a wide range of homeotic transformations
[40]. Interestingly,
KMT2A was demonstrated as having an important role in the maintenance of memory Th2 cell function
[41] and in hematopoiesis, as its absence caused defects both in self-renewal of murine hematopoietic stem cells and in hematopoietic progenitor cell differentiation in zebrafish
[42][43]. In addition, impairments in neural development were observed knocking down
Kmt2a in zebrafish, and in murine models
Mll1 was identified as a crucial component in memory formation, complex behaviors and synaptic plasticity
[44][45][46][47][48].
Thus,
KMT2A-depleted animal models recapitulate phenotypes described for patients with both germline and somatic mutations.
KMT2A associated syndromes show clinical signs such as ID, behavioral problems, speech and growth delay and peculiar dysmorphisms, while the most frequent tumors enriched in
KMT2A mutations are the hematological ones (e.g., B-cell lymphoma, T-cell lymphoblastic leukemia, acute myeloid leukemia), according to neurodevelopmental and hematopoietic defects found in the aforementioned in vivo models.
5. Conclusions
Epigenetic modifications are fundamental for many biological processes; indeed, alterations of genes with this activity can lead to neurodevelopmental disorders or tumorigenesis, when germinal or somatic mutations respectively occur
[49][50]. This is the case of
KMT2A, a lysin methyltransferase-coding gene, whose variants are associated with a chromatinopathy (WDSTS) at germinal level or can be found in both blood cancers and solid tumors in regard to malignancies.
Interestingly, due to exome- and genome-wide analyses, patients described above with a defined initial chromatinopathy diagnosis but lacking the molecular one were found to be carriers of pathogenetic variants in the
KMT2A gene and could have obtained a clinical re-evaluation. Indeed, mutations in different genes involved in the regulation and maintenance of chromatin state can lead to a clinical overlapping phenotype, suggesting a common affected pathway during embryonic development and the evaluation of an expanded set of genes when investigating the molecular causes for a correct diagnosis of these syndromes.
In addition, somatic mutations in
KMT2A have been reported in different tumors, as well as alterations in all
KMT2 family genes
[51] and in other genes associated to chromatinopathies. Curiously,
itwe is observed that germline mutations described in the literature are more frequently nonsense than missense, in contrast to somatic ones. This could be explained by the consequent loss of function mechanism characterizing most of chromatinopathies due to a defective protein production, which strongly impacts on embryonic development.
To conclude, since molecular defects in KMT2A also characterize some types of tumors, and research in the field of epigenetic drugs for malignancies is rapidly evolving
[52],
a therapeutic approach targeting KMT2A interaction or its pathway could be considered also for chromatinopathies, modulating epigenetic dysfunction with pharmaceutical products or diet-based interventions.