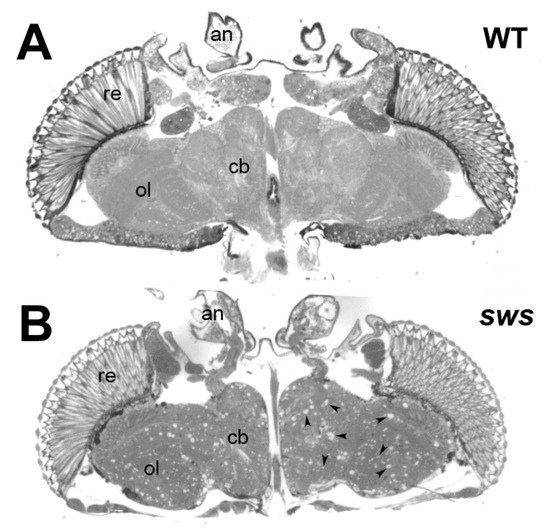
Patatin-like phospholipase domain-containing protein 6 (PNPLA6), originally called Neuropathy Target Esterase (NTE), belongs to a family of hydrolases with at least eight members in mammals. PNPLA6/NTE was first identified as a key factor in Organophosphate-induced delayed neuropathy, a degenerative syndrome that occurs after exposure to organophosphates found in pesticides and nerve agents. More recently, mutations in PNPLA6/NTE have been linked with a number of inherited diseases with diverse clinical symptoms that include spastic paraplegia, ataxia, and chorioretinal dystrophy. A conditional knockout of PNPLA6/NTE in the mouse brain results in age-related neurodegeneration, whereas a complete knockout causes lethality during embryogenesis due to defects in the development of the placenta. PNPLA6/NTE is an evolutionarily conserved protein that in Drosophila is called Swiss-Cheese (SWS). Loss of SWS in the fly also leads to locomotory defects and neuronal degeneration that progressively worsen with age.
- Neuropathy Target Esterase
- Swiss-Cheese
- organophosphate-induced delayed neuropathy
- hereditary spastic paraplegia
- phosphatidylcholine
- Protein kinase A
1. Introduction
2. Phenotypic Consequences Due to the Loss of PNPLA6/NTE in Model Systems
2.1. Drosophila Melanogaster
2.2. Danio Rerio
2.3. Mus Musculus
3. PNPLA6 in Human Disease
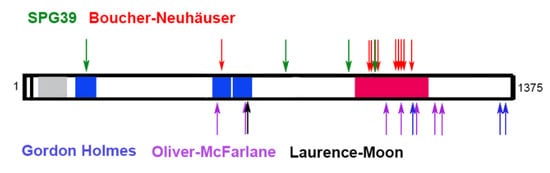
| Ataxia | Hypogonadism/ Delay in Sexual Development |
Chorioretinal Dystrophy | Trichomegaly/ Alopecia |
Spastic Paraplegia | Intellectual Disabilities | Dwarfism/ Short Statue |
|
|---|---|---|---|---|---|---|---|
| SPG 39 | +/− | + | |||||
| Boucher–Neuhäuser | + | + | +/- | +/− | +/− mild |
||
| Gordon Holmes | + | + | +/− | +/− mild |
|||
| Oliver–McFarlane | +/− | + | + severe |
+ | +/− | + | + |
| Laurence–Moon | +/− | + | + | +/− | + | + |