TLR receptors are a classic example of pattern recognition receptors (PRRs). Signals received by these receptors by recruiting specific molecules lead to activation of the transcription factors NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) and IRF (interferon regulatory factor) and affect various elements of the host’s innate immune response . TLR mechanisms are based on the ability to recognize twofold signals. The first one is based on the detection of pathogen-associated molecular patterns (PAMPs), while the second reads molecules related to damage to the body’s own cells (DAMPs; danger-associated molecular patterns). One of the major challenges faced by modern nephrology is the identification of biomarkers associated with histopathological patterns or defined pathogenic mechanisms that may assist in the non-invasive diagnosis of kidney disease, particularly glomerulopathy. The identification of such molecules may allow prognostic subgroups to be established based on the type of disease, thereby predicting response to treatment or disease relapse. Advances in understanding the pathogenesis of diseases, such as membranous nephropathy, minimal change disease, focal segmental glomerulosclerosis, IgA (immunoglobulin A) nephropathy, and diabetic nephropathy, along with the progressive development and standardization of plasma and urine proteomics techniques, have facilitated the identification of an increasing number of molecules that may be useful for these purposes.
- Toll-like receptor
- Biomarkers
- Glomeluronephritis
1. Introduction
The role of TLR receptors as potential marker molecules for the development of neoplastic diseases is emphasized more and more often, e.g., TLR5 as a prognostic marker for gastric cancer [4]. It was also shown that the activity of TLR receptor is correlated with the state of injury of post-surgical patients who have a disorder of the immune response related to the interaction of TLR receptors with DAMP (damage-associated molecular pattern). Moreover, the analyses led to conclusions regarding the role of TLR receptors in predicting pathological conditions, including tissue damage, in these patients [5].[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124]
2. Characteristic of the TLR Receptors in Glomeluronephritis
TLR receptors are a classic example of pattern recognition receptors (PRRs). Signals received by these receptors by recruiting specific molecules lead to activation of the transcription factors NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) and IRF (interferon regulatory factor) and affect various elements of the host’s innate immune response [22]. TLR mechanisms are based on the ability to recognize twofold signals. The first one is based on the detection of pathogen-associated molecular patterns (PAMPs), while the second reads molecules related to damage to the body’s own cells (DAMPs; danger-associated molecular patterns) [23]. However, the origin of the signal, in the case of PAMP, is compounds of exogenous origin, while DAMP receives endogenous information. The result of receiving signals from both pathways is the effector reaction in which the production of costimulatory molecules and cytokines takes place. The location of TLRs is the cell surface or intracellular compartments, including ER (endoplasmic reticulum), lysosomes, and endosomes. It is pointed out that intracellular localization is important not only for ligand recognition but also for avoiding TLR contact with self-nucleic acids, which could cause autoimmunity. A high number of TLR receptors on cells of the immune system, such as monocytes, macrophages, dendritic cells, or lymphocytes, enables the thesis that they constitute a network allowing rapid cooperation of leukocytes and cells present at the site of infection or in the immediate vicinity of damaged host cells [23,24][23][24]. TLR synthesis begins in the rough endoplasmic reticulum, from where it goes to the Golgi apparatus and then to its destinations, in other words to the cell surface or to the intercellular compartments [22].
At present, there are 10 types of TLR receptors in humans and three additional types in mice, whereas others species may have more of these receptors (Table 1). Based on the amino acid sequence homology, TLRs occurring in vertebrates were divided into six subfamilies: TLR 1/2/6/10, TLR3, TLR4, TLR5, followed by TLR 7/8/9, and TLR 11 to the last 12/13 (Table 2). However, not all vertebrates have all types of receptors. PRRs have a specific structure in the form of transmembrane proteins, being an integral component of the cell membrane, in which the N-terminal part is responsible for ligand binding, whereas the C-terminal end is equipped with a signaling domain for IL-1 (TIR; toll IL-1 receptor), being part of the signal induction cascade for the production of anti-inflammatory mediators [23]. The transmembrane domain of TLRs contains about 20, mostly hydrophobic, amino acid residues. The N-terminal end (ECDs (extracellular domain) N-terminal ectodomains) is a glycoprotein of 500–800 amino acid residues. In their structure, we distinguish the presence of leucine-rich tandem repeats (LRRs), the number of which depends on the receptor type and ranges from 20–29 repeats (Table 1).
Table 1. Characteristics of individual TLRs (toll-like receptors).
| Name | Location of Coding Genes | Location in the Cell |
The Number of Amino Acids | Molecular Weight (kDa) | Number of LLR | Reference |
|---|
| TLR1 | Chromosome 4 | Golgi apparatus, Phagosome, Cell membrane |
786aa | 90.31 | 19 | [25,26,27] | [25][ |
TLRs also differ in the cell site. TLR 1/2/4/5/6/10/11/12 receptors are located on the outer membrane of cells, while 3/7/8/9 receptors are located inside of them. Several literature reports have identified specific PAMP and DAMP ligands, which are bound by particular TLRs (Table 2).
Table 2. Location and ligands bound by TLR.
| Name | Occurrence | Ligand PAMP | The Origin of PAMP | Ligand DAMP | Reference |
|---|
| 26 | |||
| ] | [ | 27 | ] |
| Extracellular | ||||||||||||
| TLR2 | Chromosome 4 | Phagosome | 784aa | 89.83 | 19 | |||||||
| TLR1 | Macrophages Neutrophils B lymphocytes Dendritic cells |
Lipopeptides Soluble factors (lipoproteins) | [ | 25 | , | 28,29] | Bacteria | [ | No data | [47,48] | 25][28][29] | |
| TLR3 | Chromosome 4 | Early endosome, ER |
904aa | 103.82 | ||||||||
| TLR2 | Macrophages Neutrophils B lymphocytes Dendritic cells NK cells |
Bacterial lipopeptides Teichoic acids, | 23 | LAM Moduline, Glycolipids of bacteria, Porins, LPS, |
Bacteria | Apolipoprotein CIII, Heparin sulphate, Hyaluronic acid, Hsp60, Hsp70, Peroxiredoxin |
[47,48,49,50] | [25,30,31] | [25][30][31] | |||
| TLR4 | Chromosome 9 | Cell membrane, Early endosome | ||||||||||
| Glycosinositolphospholipids | 839aa | 95.68 | 21 | [ | 25 | ,32,33] | [25] | |||||
| Protozoa, e.g., Trypanosoma cruzi | TLR5 | Chromosome 1 | No data | 858aa | 97.83 | |||||||
| Zymosan | 20 | Fungi | [ | 25 | , | 34,35] | [25] | |||||
| TLR6 | Chromosome 4 | Golgi apparatus, Cell membrane, Phagosome |
796aa | 91.88 | ||||||||
| Hemagglutinin | 19 | [ | 25 | , | 36 | , | Measles virus37] | [25] | ||||
| TLR7 | Chromosome X | Endosomes, Lysosomes, ER, Phagosome |
1049aa | 120.92 | 25 | [25,38] | [25] | |||||
| Protein | Herpesvirus | TLR8 | Chromosome X | No data | 1041aa | 119.82 | 25 | [25,39,40] | [25] | |||
| Hsp70 proteins | Host organism | TLR9 | Chromosome 3 | Endosomes, | ||||||||
| TLR4 | Macrophages Neutrophils B lymphocytes Dendritic cells NK cells | Lysosomes, ER, Phagosome |
1032aa | Treg cells | 115.86 | LPS | Bacteria | C-reactive protein, Fibronectin, Fibrinogen, Heparin sulphate, Neutrophil, Elastase, Angiotensin II, | 25 | Hsp60 | [25,41] | [25] |
| TLR10 | Chromosome 4 | No data | 811aa | 94.56 | 19 | [25,42,43] | [25] | |||||
| TLR11 | Expression in mice | No data | 926aa | 105.83 | 10 | [44] | ||||||
| [ | TLR12 | Expression in mice | No data | 906aa | 99.94 | 17 | [45] | |||||
| 48 | , | 49 | , | 50 | ,51] | |||||||
| Fusion proteins, Proteins present in the coating |
Viruses, e.g., RSV virus | |||||||||||
| Taxol | Plants | |||||||||||
| Hsp60 protein Hsp70 protein A fragment of the A domain of fibronectin Hyaluronic acid oligosaccharide Fibrinogen Heparan sulphate |
Host organism | TLR13 | Expression in mice | Endosomes | 991aa | 114.44 | 25 | |||||
| TLR5 | Macrophages B lymphocytes Dendritic cells Treg cells |
Flagellin | Bacteria (Gram-negative) | [ | 46 | ] | ||||||
| No data | [ | 48 | , | 52 | ] | |||||||
| TLR6 | Macrophages Neutrophils Dendritic cells |
Diacyl lipopeptides Lipoteichoic acids Zymosan |
Bacteria Fungi |
Versican | [47,48,49,50,53] | |||||||
| TLR10 | Dendritic cells | No data | No data | No data | [54] | |||||||
| TLR11 | Macrophages Dendritic cells |
Flagellin Profilin |
Bacteria Protozoa, e.g., Toxoplasma gondii |
No data | [55,56] | |||||||
| TLR12 | Dendritic cells | Profilin | Protozoa, e.g., Toxoplasma gondii |
No data | [55] | |||||||
| Intracellular | ||||||||||||
| TLR3 | Macrophages Neutrophils B lymphocytes Dendritic cells |
Double-stranded RNA | Viruses | Own double-stranded RNA | [57,58] | |||||||
| TLR7 | Macrophages Neutrophils Dendritic cells Treg cells |
Single-stranded RNA Antiviral and anticancer compounds |
Viruses Synthetic |
Own single-stranded RNA | [48,59] | |||||||
| TLR8 | Dendritic cells Treg cells |
Single-stranded RNA Antiviral compounds |
Viruses Synthetic |
Own single-stranded RNA | [48,60] | |||||||
| TLR9 | Macrophages Neutrophils Dendritic cells |
Double-stranded DNA (containing unmethylated CpG sequences) | Bacteria, viruses and synthetic | HMGB1 Mitochondrial DNA |
[48,49,50,61] | |||||||
2. The Role of TLRs in Glomerulonephritis
Glomerulonephritis is a heterogeneous group of diseases whose common denominator is inflammation, ongoing in the glomerulus, resulting from systemic (secondary glomerulonephritis) or only glomerulonephritis (primary glomerulonephritis) [14,15,16,17,18,19]. The etiopathogenesis and the cause of the variable course of glomerulopathy is the subject of numerous studies but remains unknown. Although the pathogenesis is not unequivocally elucidated, the literature data clearly indicate the involvement of various immune mechanisms in the etiopathogenesis of glomerulopathy. Researchers indicate the role of the immune system in the development of chronic kidney disease on the basis of primary and secondary disorders of the glomerular functions, and AKI (acute kidney injury) resulting from the above-mentioned entities and other disease states, e.g., sepsis [20]. The main elements involved in promoting kidney damage are dendritic cells, NK (natural killer) cells, macrophages, and proinflammatory cytokines. A critical role is played by the complement system, which can both protect and promote damage to the glomeruli [21].
Tissue damage glomerulonephritis predisposes many factors and individual characteristics. Most often, the first reveals hereditary predispositions in response to emerging environmental factors that can lead to a nephrogenic immune response. Then, there is a direct exposure to infectious etiological factors occurring in the environment (PAMP and DAMP), which may be subject to specific modification due to many epigenetic factors (such as physical exercise, microbes, environmental toxins, lifestyle, etc.) [62]. As a consequence of these changes, the innate immune system is activated as a result of interaction with TLRs present on circulating inflammatory cells (neutrophils, macrophages, basophils, and NK cells), as well as on resident glomerular cells and the complement system, which triggers a cascade of antigen-specific non-specific reactions [63]. These receptors are displayed on cells found inside the glomerulus (mesangial cells, monocytes, or dendritic cells) as well as in the renal interstitium (tubular epithelial cells, monocytes), where they interfere with potential ligands [63]. Part of the ligands, such as peptides, structural elements, or genetic material of both bacteria and viruses can be transmitted through the bloodstream to the inside of the nephron, in particular to the glomerulus. In the case of interstitial cells, in addition to the ligands of infectious origin, the potential ligands may also be fibrinogen, fibronectin, defensin 2, or necrotic cells (Figure 1).
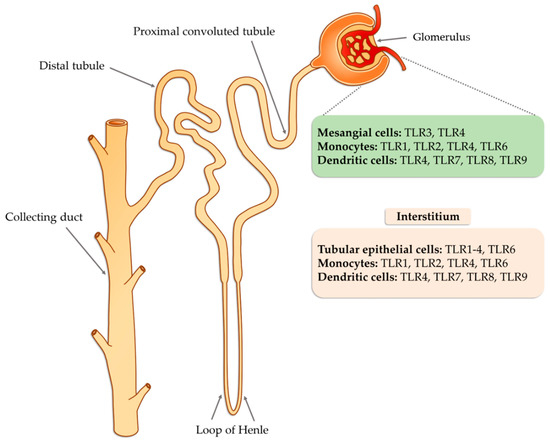
Figure 1. The potential occurrence of TLR receptors within the nephron (modified, based on [63]).
In order to prevent over-activity of the immune system, TLRs are downregulated by numerous molecules and various mechanisms. Existing negative regulators target specific key molecules in TLR signaling, such as SOCS1 (suppressor of cytokine signaling 1), SOCS3 (suppressor of cytokine signaling 3), SARM (sterile α-and armadillo-motif containing protein), TANK (TRAF family member associated NF-κB activator), A20, and others [64,65,66,67,68,69]. Additionally, there are molecules that directly influence the inhibition of NF-kB and IRF-3 [70]. In addition, numerous mi-RNAs were discovered that affect the stability of mRNA encoding signaling molecules and mRNA for cytokines [69,71].
Activation of TLR releases the NF-κB transcription factor, which results in the production of inflammatory mediators (such as IL-1, IL-2, IL-6, IL-12, TNF-α (tumor necrosis factor alpha)), [72,73], and can cause glomerular damage. The next step is the conversion of the innate immune response that begins the antigen-specific reaction cycle. The transformation of the immune response includes several possible mechanisms, such as regulation of natural autoimmunity, conformational changes of epitopes, molecular mimicry, or the autoantigen complementarity phenomenon. TLRs are also required to activate the adaptive immune system by antigen-presenting cells that promote CD4 helper cell differentiation, B cell activation, and antibody production. Antibodies lead to the trapping of the circulating complex or the formation of in situ immune complexes that can activate both TLRs and complement components of the innate immune system [74]. CD4 Th1 and Th2 cells cause damage to the glomerular tissues indirectly, mainly through macrophages and basophils, whereas Th17 cells may directly mediate damage to kidney structures in particular diseases (Figure 2).
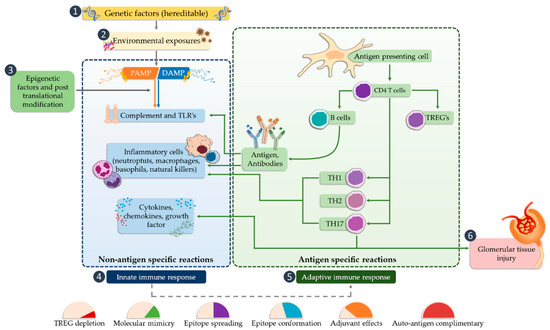
Figure 2. The potential occurrence of TLR receptors within the nephron (modified, based on [24]).
Various clinical situations affecting the kidneys, such as ischemic damage, toxic AKI, nephropathies secondary to diabetes mellitus, hypertension, or crystal deposition, are associated with aseptic inflammation caused, among others, by DAMP molecules [75,76]. These molecules can be released from dying parenchyma cells or during remodulation of the extracellular matrix. The presence of cells in the kidney capable of expressing TLR receptors makes it possible to initiate an immune response and inflammation [77,78]. In order to approximate the mode of action of these receptors in the disease state, their involvement in the most frequently diagnosed pathological conditions associated with glomerular dysfunction is presented in the further part of the material [8].
3. Biomarkers and Importance of the TLRs in Selected Glomerular Diseases
The literature classification divides the occurring nephropathies into two categories: Primary nephropathies, which are defined as those in which the systemic disease responsible for the condition cannot be established, and secondary nephropathies in which renal lesions appear as a result of other diseases accompanied by characteristic extrarenal symptoms. The first group includes diseases, such as: minimal change nephropathy (MCN), focal segmental glomerulosclerosis (FSGS), and membranous nephropathy (MN), which are also included in the nephrotic syndrome. The second group is primarily diabetic nephropathy and lupus nephropathy. In addition, the studies of our research group have resulted in the finding that the TLR-2 receptor may play an important role as a biomarker of primary non-proliferative nephropathies [79].
4. The Role of TLRs in Primary Nephropathies
4.1. Focal Segmental Glomerulosclerosis (FSGS)
FSGS is a diverse syndrome that arises after damage to podocytes for various reasons, some known and unknown. The sources of podocyte injury are diverse (circulating factors (primary FSGS), genetic abnormalities, viral infections, and medications) [80,81]. Most of the mutual interactions between these factors probably result in FSGS. There is a hypothesis about multistage pathogenic activation of autoimmunity in some forms of idiopathic FSGS [81]. Through the interaction of macrophages involved in kidney damage with many chemokines, the migration of monocytes to the site of damage occurs, which initiates the process of fibrosis. These macrophages also have the ability to self-spread and change into myofibroblasts that produce the extracellular matrix. Therefore, it can be assumed that excessive organ infiltration by monocytes and macrophages will cause an intensified fibrosis effect and, consequently, intensification of FSGS symptoms [82,83]. Currently, known biomarkers of FSGS are soluble urokinase-type plasminogen activator receptor (suPAR), soluble IL-2 receptor (sIL-2R), and ATP-binding cassette subfamily B member 1 glycoprotein-P (Figure 3). Damage to podocytes can release molecular patterns of proteins that are recognized by TLR as signals of danger. TLRs stimulate adapter proteins that activate a cascade of kinases, which amplify the signal and transmit it to the transcription factors regulating inflammatory genes. In the inflammatory microenvironment, the podocytes, acting as antigen-presenting cells, have a CD40 and CD80 receptor on their surface, thanks to which they capture antigens and present them to competent T-cells. However, in the case of abnormal expression of CD40 and CD80, they disorganize the cytoskeleton and filtration slit. In addition, CD40 can be identified as a foreign antigen, consequently leading to the production of anti-CD40 auto-antigen. Abnormal expression of CD40, CD80, and autoantibodies may lead to apoptosis of the podocytes, detachment of the podocytes from the glomerular basal membrane, proliferation of parietal epithelial cells, and attack on the glomeruli, and induction of segmental sclerosis [81]. In turn, other literature reports indicate the involvement of fibrinogen (Fg) in an inflammatory process mediated by the toll-like 4 receptor (TLR4) [84]. Fibrinogen is a protein that plays a proinflammatory role in vascular disorders, rheumatoid arthritis, glomerulonephritis, and certain cancers, e.g., myeloproliferative neoplasms [84,85]. Positive correlations have been noted between oxidative stress markers and, among others, fibrinogen, which may impact the course of several disorders [85]. Găman et al. [86] showed that obesity and diabetes are associated with increased levels of ROS (reactive oxygen species), accompanied by a simultaneous deficiency of antioxidants. The authors showed that the results of the free oxygen radical defense (FORT) and free oxygen radical defense (FORD) tests correlated with anthropometric/biochemical parameters in patients with obesity and diabetes. In studies carried out by Wang et al. [84], Fg has been shown to disrupt the actin cytoskeleton and induce apoptosis in podocytes via the TLR4-p38 MAPK-NF-κB p65 pathway in vitro and that co-expression of Fg and TLR4 is elevated in podocytes of Adriamycin-treated mice. It was also indicated that the level of fibrinogen in the urine may reflect the disease activity in patients with FSGS [84]. Literature data show that the use of synthetic small molecules lecinoxoids, which are inhibitors of TLR-2 and TLR-4, affects the activation and recruitment of monocytes in a rat model. The authors indicate [87] that the data demonstrate that targeting TLR-2-TLR-4 and/or monocyte migration directly affects the priming phase of fibrosis and may consequently perturb disease pathogenesis.
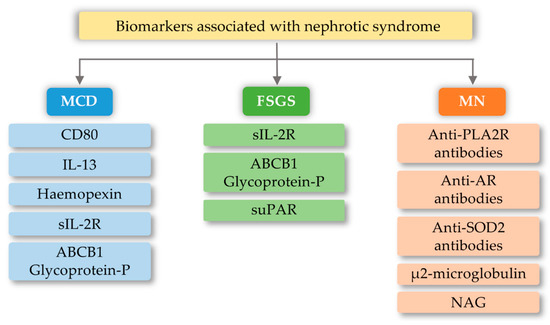
Figure 3. Biomarkers associated with nephrotic syndrome based on minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), and membranous nephropathy (MN) change based on [88]). CD80-Cluster of differentiation 80; IL-13-interleukin 13; sIL-2R-soluble IL-2 receptor; ABCB1 Glycoprotein-P-ATP-binding cassette subfamily B member 1 Glycoprotein-P; suPAR-soluble urokinase-type plasminogen activator receptor; PLA2R-M-type phospholipase A2 receptor; SOD2-manganese superoxide-dismutase 2; AR-Androgen Receptor; NAG-N-Acetyl-β-D Glucosaminidase.
4.2. Minimal Change Disease (MCD)
MCD is one of the most common glomerular kidney diseases in children and a common cause of nephrotic syndrome in adults. This disease entity is characterized by an outbreak of edema, selective proteinuria, and a clinical response to glucocorticoid therapy, as T-cell mechanisms are involved in the pathogenesis of the disease [89]. The pathogenesis is due to abnormalities in the functioning of podocytes, with the latest literature data suggesting the hypothesis that there are two initiating events. First, there are changes in the cytoskeleton of podocytes and, second, there are regulatory changes in T-cells that exacerbate abnormalities in podocytes [89,90]. Currently known biomarkers in MCD are urine levels and podocyte expression of CD80 (B7.1), interleukin 13, serum levels and protease activity of circulating hemopexin, serum levels of soluble interleukin 2 receptor, and ABCB1 and glycoprotein-P (Figure 3). The development of MCD may be significantly influenced by the body’s innate immunity, in which TLRs are involved. Podocytes in the kidney glomeruli, due to their function and place of occurrence, are also equipped with the above receptors. In research conducted by Srivastava et al. [91], the presence of TLR receptors and their potential activity were checked on cell cultures stimulated with LPS (lipopolysaccharides) and the amino nucleoside puromycin (PAN). In the above studies, it was found that cultured human podocytes constitutively express TLR 1-6 and TLR-10 but not TLR 7–9. Quantitative analysis using the RT-PCR method indicated that LPS at various concentrations and to varying degrees increased the expression of TLR (1–6) genes, the adapter molecule MyD88, and the transcription factor NF-κB within one hour. LPS also caused elevated levels of IL-6, IL-8, and MCP-1 (monocyte chemoattractant protein-1) without exerting any effect on TNF-α, IFN-α, or TGF-β1 after 24 h. It has also been shown that an increase in TLR 1 expression may attenuate the effect of TLR-4 activation, which is thought to be an indirect factor in LPS-induced podocyte damage [91]. Moreover, the increase in TLR-1 expression by LPS suggests that LPS damage to podocytes is associated with an increase in TLR-1 levels and its specific endogenous ligands (heat shock protein, heparin sulphate, and fibronectin). These results allow the conclusion that the main TLR4 ligand, which is LPS, can induce the expression of the genes of many TLR receptors, and thus may lead to changes being induced in podocytes, which may be related to the loss of receptor selectivity and stimulation of receptor interaction in podocytes [91]. An additional possibility indicating the involvement of TLRs in the development of MCD is the increase in the amount of the CD80 receptor in podocytes, after stimulation with ligands for TLR-and TLR-4 receptors. TLR ligands are usually microbial products and can be combined with a well-known association of viral infections as a causative agent of minimal lesion disease [92,93].
4.3. Membranous Nephropathy (MN)
MN is a common cause of nephrotic syndrome in adults. Patients with MN usually develop severe proteinuria, edema, hypoalbuminemia, and hyperlipidemia [94]. It is the most common cause of idiopathic nephrotic syndrome in non-diabetic white adults. About 80% of cases are restricted to the kidneys (primary MN, PMN, idiopathic membranous nephropathy) and 20% are related to other systemic diseases or exposure (secondary MN) [95]. MN is associated with a pathological alteration of the glomerular basement membrane. This change is due to the build-up of immune complexes that appear as granular immunoglobulin (Ig)G deposits after immunofluorescence imaging and as electron-dense deposits of high electron density. Deposits of these immune complexes between podocytes and the basement membrane have a complex that attacks the complement membrane (C5b-9) [96]. The formation of glomerular sub-epithelial immune complex deposits in the IMN is mediated by specific intrinsic podocyte antigens and their corresponding autoantibodies in humans. These include compounds, such as neutral endopeptidase (NEP), type M receptor for secretory phospholipase A2 (PLA2R1), and type 1 7A thrombospondin (THSD7A) (containing domain 8–10) [94,95,96]. The above-mentioned markers constitute the core of the research into the pathogenesis of membranous nephropathy. However, there are reports of a genetic susceptibility to idiopathic membranous nephropathy. This type of study was conducted in a high-prevalence area in Taiwan [97]. In these studies, the association of the IL-6, NPHS1 (nephrin), TLR-4, TLR-9, STAT4 (signal transducer and activator of transcription) and MYH (mutY DNA glycosylase), genes with susceptibility to primary membranous nephropathy in Taiwan was established. In the case of the TLR4 receptor gene, the gene polymorphism indicated a significant single nucleotide difference in the rs10983755 A/G region (p < 0.001) and rs1927914 A/G (p < 0.05) between the control group and MN patients. In addition, the distributions of rs10759932 C/T and rs11536889 C/T polymorphisms differed significantly [97].
4.4. IgA Nephropathy (IgAN)
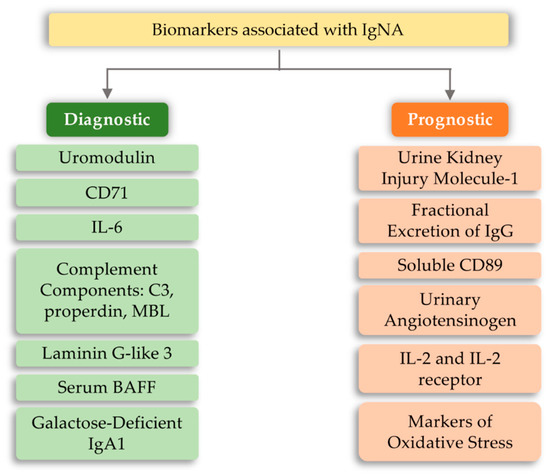
The development of IgA nephropathy consists of many mechanisms not yet fully understood. Literature data breaks down biomarkers for IgA nephropathy into a diagnostic and prognostic marker. The first group includes biomarkers detected in serum and urine, such as uromodulin, CD71, IL-6, complement components, and serum BAFF (B-cell activating factor) [98]. However, the group of prognostic markers includes urine kidney injury molecule-1, fractional excretion of IgG, soluble CD89, urinary angiotensinogen, and inflammatory cytokines (Figure 4). The above markers may indicate or predict the main cause of nephropathy development, which is the overproduction of anti-IgA complexes. One of the reasons for stimulating the body to produce IgA-related complexes may be signaling disorders associated with TLRs [99,100,101]. Ligands of bacterial or viral origin are recognized by toll-like receptors that trigger the process of chemokine release and recruitment of macrophages and neutrophils at the site of infection [102]. In numerous studies on IgAN pathogenesis, various types of receptors that could influence the development of this disease have been analyzed. These receptors were TLR3, mainly recognizing viral dsDNA [103]; TLR7 receptor [104]; binding to ssRNA viruses and TLR4 [105,106]; and binding of a variety of ligands, including LPS Gram-negative bacteria and DAMP (Table 2). Numerous observations of patients with diagnosed IgAN suggest the involvement of pathogens of viral origin, which is also confirmed by experimental models. There is an unexpected increase in the level of TLR4 activation, which is involved in the diagnosis of exogenous bacterial factors (LPS from Gram-negative bacteria, Chlamydia pneumoniae, HSP (heat shock proteins) proteins) and endogenous origin (HSP-60, additional fibronectin A domain, low-molecular LDL fractions, acid oligosaccharides hyaluronic acid, heparan sulphate), as well as factors derived from the breakdown of host cells [107,108,109,110,111,112,113]. To fully explain kidney damage in IgAN, it is necessary to fully understand the effects of TLR4 in the development of glomerulopathy, involving both glomerular cells and circulating leukocytes. Studies have shown that the administration of LPS activates TLR4 receptors on mesangial cells, and causes the release of chemokines (CXCs), which promotes neutrophil infusion and the development of glomerulonephritis [114,115,116]. In addition, the IFN-γ and IFN-α responses induced by TLR activation induce overexpression of the B-cell activation factor (BAFF) in dendritic cells, favoring the expansion of B-cells and increasing IgA synthesis [117,118,119,120,121]. It was also shown that in kidney biopsies of patients with IgAN, CD19+/CD5+ B cell infiltration is present, which in the progressive forms of this disease produce significant amounts of IFN-γ and IgA and are more resistant to apoptosis compared to cells obtained from healthy donors [122,123]. Moreover, Hitoshi Suzuki et al. [101] showed that there is an association between gene polymorphisms for TLR-9 and disease progression. Stimulation with ligands for TLR-9 led to the deterioration of kidney function in mice and influenced the shift of the balance towards Th1 lymphocytes. These findings led to the conclusion that activation of pathways related to this particular type of receptor may influence the severity of IgA nephropathy [101]. Moreover, Coppo et al. [124] showed that in patients diagnosed with IgA nephropathy, higher levels of TLR-4 in mononuclear cells and transcriptional mRNA were observed than in the control group. An important fact is that there is a statistical difference in the level of the above markers in patients with severe disease and those who do not have proteinuria and hematuria [124]. TLR-4 can be activated by many ligands, such as HLPs and LPS and DAMPs [106].
References
- World Health Organization; International Programme on Chemical Safety. Biomarkers in Risk Assessment: Validity and Validation; World Health Organization: Geneva, Switzerland, 2001; ISBN 978-92-4-157222-4. [Google Scholar]
- Fuentes-Arderiu, X. What is a biomarker? It’s time for a renewed definition. Clin. Chem. Lab. Med. 2013, 51, 1689–1690. [Google Scholar] [CrossRef] [PubMed]
- Goerlich, N.; Brand, H.A.; Langhans, V.; Tesch, S.; Schachtner, T.; Koch, B.; Paliege, A.; Schneider, W.; Grützkau, A.; Reinke, P.; et al. Kidney transplant monitoring by urinary flow cytometry: Biomarker combination of T cells, renal tubular epithelial cells, and podocalyxin-positive cells detects rejection. Sci. Rep. 2020, 10, 796. [Google Scholar] [CrossRef] [PubMed]
- Kasurinen, A.; Hagström, J.; Laitinen, A.; Kokkola, A.; Böckelman, C.; Haglund, C. Evaluation of toll-like receptors as prognostic biomarkers in gastric cancer: High tissue TLR5 predicts a better outcome. Sci. Rep. 2019, 9, 12553. [Google Scholar] [CrossRef] [PubMed]
- Darrabie, M.D.; Cheeseman, J.; Limkakeng, A.T.; Borawski, J.; Sullenger, B.A.; Elster, E.A.; Kirk, A.D.; Lee, J. Toll-like receptor activation as a biomarker in traumatically injured patients. J. Surg. Res. 2018, 231, 270–277. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Dobrică, E.-C.; Găman, M.-A.; Cozma, M.-A.; Bratu, O.G.; Pantea Stoian, A.; Diaconu, C.C. Polypharmacy in Type 2 Diabetes Mellitus: Insights from an Internal Medicine Department. Medicina 2019, 55, 436. [Google Scholar] [CrossRef]
- O’Shaughnessy, M.M.; Hogan, S.L.; Thompson, B.D.; Coppo, R.; Fogo, A.B.; Jennette, J.C. Glomerular disease frequencies by race, sex and region: Results from the International Kidney Biopsy Survey. Nephrol. Dial. Transplant. 2018, 33, 661–669. [Google Scholar] [CrossRef]
- Naylor, S. Biomarkers: Current perspectives and future prospects. Expert Rev. Mol. Diagn. 2003, 3, 525–529. [Google Scholar] [CrossRef]
- Sahu, P.; Pinkalwar, N.; Dubey, R.D.; Paroha, S.; Chatterjee, S.; Chatterjee, T. Biomarkers: An Emerging Tool for Diagnosis of a Disease and Drug Development. Asian J. Res. Pharm. Sci. 2011, 1, 9–16. [Google Scholar]
- Drucker, E.; Krapfenbauer, K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J. 2013, 4, 7. [Google Scholar] [CrossRef]
- Mayeux, R. Biomarkers: Potential Uses and Limitations. NeuroRx 2004, 1, 182–188. [Google Scholar] [CrossRef] [PubMed]
- FDA. About Biomarkers and Qualification. Available online: https://www.fda.gov/drugs/cder-biomarker-qualification-program/about-biomarkers-and-qualification (accessed on 10 August 2020).
- Levin, A.; Tonelli, M.; Bonventre, J.; Coresh, J.; Donner, J.-A.; Fogo, A.B.; Fox, C.S.; Gansevoort, R.T.; Heerspink, H.J.L.; Jardine, M.; et al. Global kidney health 2017 and beyond: A roadmap for closing gaps in care, research, and policy. Lancet 2017, 390, 1888–1917. [Google Scholar] [CrossRef]
- Devarapu, S.K.; Anders, H. Toll-like receptors in lupus nephritis. J. Biomed. Sci. 2018, 25, 35. [Google Scholar] [CrossRef]
- Luyckx, V.A.; Tonelli, M.; Stanifer, J.W. The global burden of kidney disease and the sustainable development goals. Bull. World Health Organ. 2018, 96, 414–422D. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.C.; Cheung, C.K.; Barratt, J. New insights into the pathogenesis of IgA nephropathy. Pediatr. Nephrol. 2018, 33, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zheng, F. Immune Cells and Inflammation in Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 1841690. [Google Scholar] [CrossRef]
- Mohammad Hosseini, A.; Majidi, J.; Baradaran, B.; Yousefi, M. Toll-Like Receptors in the Pathogenesis of Autoimmune Diseases. Adv. Pharm. Bull. 2015, 5, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Ryan, M.J. Immune and Inflammatory Role in Renal Disease. Compr. Physiol. 2013, 3, 957–976. [Google Scholar] [CrossRef]
- Berger, S.P.; Daha, M.R. Complement in glomerular injury. Semin. Immunopathol. 2007, 29, 375–384. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Lawson, B.R. Toll-like receptors and kidney diseases. Inflamm. Allergy Drug Targets 2009, 8, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xue, G.; Li, S.; Fu, Y.; Yin, J.; Zhang, R.; Li, J. Effect of Intermittent and Mild Cold Stimulation on the Immune Function of Bursa in Broilers. Animals 2020, 10, 1275. [Google Scholar] [CrossRef] [PubMed]
- Botos, I.; Segal, D.M.; Davies, D.R. The Structural Biology of Toll-Like Receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef] [PubMed]
- UniProt. Toll-Like Receptor 1. Available online: https://www.uniprot.org/uniprot/Q15399 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of the TLR1-TLR2 Heterodimer Induced by Binding of a tri-Acylatedlipopeptide. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/2z7x (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 2. Available online: https://www.uniprot.org/uniprot/O60603 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of TLR2-TLR6-Pam2CSK4 Complex. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/3a79 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 3. Available online: https://www.uniprot.org/uniprot/O15455 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of the Complex of TLR3 and bi-Specific Diabody. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/5gs0 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 4. Available online: https://www.uniprot.org/uniprot/O00206 (accessed on 10 August 2020).
- Protein Data Bank in Europe. The Crystal Structure of Mouse TLR4/MD-2/neoseptin-3. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/5ijc (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 5. Available online: https://www.uniprot.org/uniprot/O60602 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Homology Model of Human Toll-Like Receptor 5 Fitted into an Electron Microscopy Single Particle Reconstruction. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/3j0a (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 6. Available online: https://www.uniprot.org/uniprot/Q9Y2C9 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of TIR Domain TLR6. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/4om7 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 7. Available online: https://www.uniprot.org/uniprot/Q9NYK1 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 8. Available online: https://www.uniprot.org/uniprot/Q9NR97 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of Human TLR8 in Complex with XG-1-236. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/4qc0 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 9. Available online: https://www.uniprot.org/uniprot/Q9NR96 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 10. Available online: https://www.uniprot.org/uniprot/Q9BXR5 (accessed on 10 August 2020).
- Protein Data Bank in Europe. The TIR Domain of Human Toll-Like Receptor 10 (TLR10). Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/2j67 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 11. Available online: https://www.uniprot.org/uniprot/Q6R5P0 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 12. Available online: https://www.uniprot.org/uniprot/Q6QNU9 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 13. Available online: https://www.uniprot.org/uniprot/Q6R5N8 (accessed on 10 August 2020).
- Farhat, K.; Riekenberg, S.; Heine, H.; Debarry, J.; Lang, R.; Mages, J.; Buwitt-Beckmann, U.; Röschmann, K.; Jung, G.; Wiesmüller, K.-H.; et al. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J. Leukoc. Biol. 2008, 83, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Zhang, Q.; Wang, F.; Zhang, D. Toll-like receptors and prostate cancer. Front. Immunol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Goulopoulou, S.; McCarthy, C.G.; Clinton Webb, R. Toll-like receptors in the vascular system: Sensing the dangers within. Pharmacol. Rev. 2016, 68, 142–167. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef]
- Miao, E.A.; Andersen-Nissen, E.; Warren, S.E.; Aderem, A. TLR5 and Ipaf: Dual sensors of bacterial flagellin in the innate immune system. Semin. Immunopathol. 2007, 29, 275–288. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kawai, T.; Sanjo, H.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Takeda, K.; Akira, S. TLR6: A novel member of an expanding Toll-like receptor family. Gene 1999, 231, 59–65. [Google Scholar] [CrossRef]
- Oosting, M.; Cheng, S.C.; Bolscher, J.M.; Vestering-Stenger, R.; Plantinga, T.S.; Verschueren, I.C.; Arts, P.; Garritsen, A.; van Eenennaam, H.; Sturm, P.; et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc. Natl. Acad. Sci. USA 2014, 111, E4478–E4484. [Google Scholar] [CrossRef] [PubMed]
- Raetz, M.; Kibardin, A.; Sturge, C.R.; Pifer, R.; Li, H.; Burstein, E.; Ozato, K.; Larin, S.; Yarovinsky, F. Cooperation of TLR12 and TLR11 in the IRF8-Dependent IL-12 Response to Toxoplasma gondii Profilin. J. Immunol. 2013, 191, 4818–4827. [Google Scholar] [CrossRef] [PubMed]
- Yarovinsky, F.; Zhang, D.; Andersen, J.F.; Bannenberg, G.L.; Serhan, C.N.; Hayden, M.S.; Hieny, S.; Sutterwala, F.S.; Flavell, R.A.; Ghosh, S.; et al. Immunology: TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 2005, 308, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Zhao, P.; Rodriguez-Pinto, D.; Qi, D.; Henegariu, O.; Alexopoulou, L.; Flavell, R.A.; Wong, F.S.; Wen, L. Inflammatory Regulation by TLR3 in Acute Hepatitis. J. Immunol. 2009, 183, 3712–3719. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Osawa, T.; Ogami, K.; Ouyang, X.; Sakaguchi, S.; Koshiba, R.; Yanai, H.; Seko, Y.; Shitara, H.; Bishop, K.; et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2008, 105, 20446–20451. [Google Scholar] [CrossRef]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small-antiviral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Guo, Z.; Kiniwa, Y.; Voo, K.S.; Peng, W.; Fu, T.; Wang, D.Y.; Li, Y.; Wang, H.Y.; Wang, R.F. Immunology: Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 2005, 309, 1380–1384. [Google Scholar] [CrossRef]
- Notley, C.A.; Jordan, C.K.; McGovern, J.L.; Brown, M.A.; Ehrenstein, M.R. DNA methylation governs the dynamic regulation of inflammation by apoptotic cells during efferocytosis. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef]
- Anders, H.-J.; Banas, B.; Schlöndorff, D. Signaling Danger: Toll-Like Receptors and their Potential Roles in Kidney Disease. J. Am. Soc. Nephrol. 2004, 15, 854–867. [Google Scholar] [CrossRef]
- Verstak, B.; Stack, J.; Ve, T.; Mangan, M.; Hjerrild, K.; Jeon, J.; Stahl, R.; Latz, E.; Gay, N.; Kobe, B.; et al. The TLR signaling adaptor TRAM interacts with TRAF6 to mediate activation of the inflammatory response by TLR4. J. Leukoc. Biol. 2014, 96, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A deubiquitinase that regulates type I interferon production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Jin, J.; Xu, S.; Liu, H.; Li, N.; Cao, X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 2010, 11, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Skaug, B.; Chen, J.; Du, F.; He, J.; Ma, A.; Chen, Z.J. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol. Cell 2011, 44, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Kim, J.J.; Kim, S.W.; Jin, H.S.; Yang, C.S.; Park, K.A.; Chanda, D.; Kim, D.K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011, 12, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kawai, T.; Akira, S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012, 33, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Tun-Kyi, A.; Ryo, A.; Yamamoto, M.; Finn, G.; Fujita, T.; Akira, S.; Yamamoto, N.; Lu, K.P.; Yamaoka, S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 2006, 7, 598–605. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Couser, W.G.; Johnson, R.J. The etiology of glomerulonephritis: Roles of infection and autoimmunity. Kidney Int. 2014, 86, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Kitaura, J.; Izawa, K.; Kaitani, A.; Komeno, Y.; Nakamura, M.; Yamazaki, S.; Enomoto, Y.; Oki, T.; Akiba, H.; et al. TIM1 is an endogenous ligand for LMIR5/CD300b: LMIR5 deficiency ameliorates mouse kidney ischemia/reperfusion injury. J. Exp. Med. 2010, 207, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J. Toll-Like Receptors and Danger Signaling in Kidney Injury. J. Am. Soc. Nephrol. 2010, 21, 1270–1274. [Google Scholar] [CrossRef]
- Rosin, D.L.; Okusa, M.D. Dangers Within: DAMP Responses to Damage and Cell Death in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Mertowski, S.; Grywalska, E.; Gosik, K.; Smarz-Widelska, I.; Hymos, A.; Dworacki, G.; Niedźwiedzka-Rystwej, P.; Drop, B.; Roliński, J.; Załuska, W. TLR2 Expression on Select Lymphocyte Subsets as a New Marker in Glomerulonephritis. J. Clin. Med. 2020, 9, 541. [Google Scholar] [CrossRef]
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- Reggiani, F.; Ponticelli, C. Focal segmental glomerular sclerosis: Do not overlook the role of immune response. J. Nephrol. 2016, 29, 525–534. [Google Scholar] [CrossRef]
- Eardley, K.S.; Kubal, C.; Zehnder, D.; Quinkler, M.; Lepenies, J.; Savage, C.O.; Howie, A.J.; Kaur, K.; Cooper, M.S.; Adu, D.; et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008, 74, 495–504. [Google Scholar] [CrossRef]
- Wu, X.; Dolecki, G.J.; Sherry, B.; Zagorski, J.; Lefkowith, J.B. Chemokines are expressed in a myeloid cell-dependent fashion and mediate distinct functions in immune complex glomerulonephritis in rat. J. Immunol. 1997, 158, 3917–3924. [Google Scholar]
- Wang, H.; Zheng, C.; Xu, X.; Zhao, Y.; Lu, Y.; Liu, Z. Fibrinogen links podocyte injury with Toll-like receptor 4 and is associated with disease activity in FSGS patients. Nephrology 2018, 23, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Gaman, A.M.; Moisa, C.; Diaconu, C.C.; Gaman, M.A. Crosstalk between Oxidative Stress, Chronic Inflammation and Disease Progression in Essential Thrombocythemia. Rev. Chim. 2019, 70, 3486–3489. [Google Scholar] [CrossRef]
- Găman, M.A.; Epîngeac, M.E.; Diaconu, C.C.; Găman, A.M. Evaluation of oxidative stress levels in obesity and diabetes by the free oxygen radical test and free oxygen radical defence assays and correlations with anthropometric and laboratory parameters. World J. Diabetes 2020, 11, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Yacov, N.; Feldman, B.; Volkov, A.; Ishai, E.; Breitbart, E.; Mendel, I. Treatment with lecinoxoids attenuates focal and segmental glomerulosclerosis development in nephrectomized rats. Basic Clin. Pharmacol. Toxicol. 2019, 124, 131–143. [Google Scholar] [CrossRef]
- Segarra-Medrano, A.; Carnicer-Cáceres, C.; Arbós-Via, M.A.; Quiles-Pérez, M.T.; Agraz-Pamplona, I.; Ostos-Roldán, E. Biological markers of nephrotic syndrome: A few steps forward in the long way. Nefrología 2012, 32, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.R.; Choi, M. Minimal Change Disease in Adults. In Glomerulonephritis; Trachtman, H., Herlitz, L.C., Lerma, E.V., Hogan, J.J., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 97–114. ISBN 978-3-319-49379-4. [Google Scholar]
- Uwaezuoke, S.N. Biomarkers of Common Childhood Renal Diseases. In Biomarker—Indicator of Abnormal Physiological Process; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Srivastava, T.; Sharma, M.; Yew, K.H.; Sharma, R.; Duncan, R.S.; Saleem, M.A.; McCarthy, E.T.; Kats, A.; Cudmore, P.A.; Alon, U.S.; et al. LPS and PAN-induced podocyte injury in an in vitro model of minimal change disease: Changes in TLR profile. J. Cell Commun. Signal. 2013, 7, 49–60. [Google Scholar] [CrossRef]
- Mishra, O.P.; Kumar, R.; Narayan, G.; Srivastava, P.; Abhinay, A.; Prasad, R.; Singh, A.; Batra, V.V. Toll-like receptor 3 (TLR-3), TLR-4 and CD80 expression in peripheral blood mononuclear cells and urinary CD80 levels in children with idiopathic nephrotic syndrome. Pediatr. Nephrol. 2017, 32, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Shimada, M.; Araya, C.E.; Huskey, J.; Garin, E.H.; Johnson, R.J. Minimal Change Disease: A CD80 podocytopathy? Semin. Nephrol. 2011, 31, 320–325. [Google Scholar] [CrossRef]
- Lai, W.L.; Yeh, T.H.; Chen, P.M.; Chan, C.K.; Chiang, W.C.; Chen, Y.M.; Wu, K.D.; Tsai, T.J. Membranous nephropathy: A review on the pathogenesis, diagnosis, and treatment. J. Formos. Med. Assoc. 2015, 114, 102–111. [Google Scholar] [CrossRef]
- Couser, W.G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 983–997. [Google Scholar] [CrossRef]
- Liu, W.; Gao, C.; Dai, H.; Zheng, Y.; Dong, Z.; Gao, Y.; Liu, F.; Zhang, Z.; Liu, Z.; Liu, W.; et al. Immunological Pathogenesis of Membranous Nephropathy: Focus on PLA2R1 and Its Role. Front. Immunol. 2019, 10, 1809. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Chen, C.H.; Huang, Y.C.; Chan, C.J.; Chen, D.C.; Tsai, F.J. Genetic susceptibility to idiopathic membranous nephropathy in high-prevalence Area, Taiwan. Biomedicine 2014, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Nafar, M.; Samavat, S. Biomarkers in IgA Nephropathy. In Biomarkers in Kidney Disease; Patel, V.B., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–29. ISBN 978-94-007-7743-9. [Google Scholar]
- Liu, Y.; Ma, X.; Lv, J.; Shi, S.; Liu, L.; Chen, Y.; Zhang, H. Risk factors for pregnancy outcomes in patients with IgA nephropathy: A matched cohort study. Am. J. Kidney Dis. 2014, 64, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R.; Camilla, R.; Amore, A.; Peruzzi, L. Oxidative Stress in IgA Nephropathy. Nephron Clin. Pract. 2010, 116, c196–c199. [Google Scholar] [CrossRef]
- Suzuki, H.; Suzuki, Y.; Narita, I.; Aizawa, M.; Kihara, M.; Yamanaka, T.; Kanou, T.; Tsukaguchi, H.; Novak, J.; Horikoshi, S.; et al. Toll-Like Receptor 9 Affects Severity of IgA Nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2384–2395. [Google Scholar] [CrossRef]
- Rollino, C.; Vischini, G.; Coppo, R. IgA nephropathy and infections. J. Nephrol. 2016, 29, 463–468. [Google Scholar] [CrossRef]
- Merkle, M.; Ribeiro, A.; Köppel, S.; Pircher, J.; Mannell, H.; Roeder, M.; Wörnle, M. TLR3-dependent immune regulatory functions of human mesangial cells. Cell. Mol. Immunol. 2012, 9, 334–340. [Google Scholar] [CrossRef]
- Coppo, R.; Amore, A.; Peruzzi, L.; Vergano, L.; Camilla, R. Innate immunity and IgA nephropathy. J. Nephrol. 2010, 23, 626–632. [Google Scholar]
- Sheng, X.; Zuo, X.; Liu, X.; Zhou, Y.; Sun, X. Crosstalk between TLR4 and Notch1 signaling in the IgA nephropathy during inflammatory response. Int. Urol. Nephrol. 2018, 50, 779–785. [Google Scholar] [CrossRef]
- Lim, B.J.; Lee, D.; Hong, S.W.; Jeong, H.J. Toll-Like Receptor 4 Signaling is Involved in IgA-Stimulated Mesangial Cell Activation. Yonsei Med. J. 2011, 52, 610–615. [Google Scholar] [CrossRef]
- Chebotareva, N.; Bobkova, I.; Shilov, E. Heat shock proteins and kidney disease: Perspectives of HSP therapy. Cell Stress Chaperones 2017, 22, 319–343. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Mii, A.; Fukui, M.; Nagahama, K.; Shimizu, A.; Tsuruoka, S. IgA Nephropathy and Psoriatic Arthritis that Improved with Steroid Pulse Therapy and Mizoribine in Combination with Treatment for Chronic Tonsillitis and Epipharyngitis. Intern. Med. 2015, 54, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.J.; Lock, H.R.; Wolfs, T.G.A.M.; Buurman, W.A.; Sacks, S.H.; Robson, M.G. Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J. Am. Soc. Nephrol. 2007, 18, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Myllymäki, J.; Syrjänen, J.; Helin, H.; Pasternack, A.; Kattainen, A.; Mustonen, J. Vascular diseases and their risk factors in IgA nephropathy. Nephrol. Dial. Transplant. 2006, 21, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Feehally, J.; Barratt, J. The Genetics of IgA Nephropathy: An Overview from Western Countries. Kidney Dis. 2015, 1, 33–41. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-Like Receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Sano, N.; Kitazawa, K.; Sugisaki, T. Localization and roles of CD44, hyaluronic acid and osteopontin in IgA nephropathy. Nephron 2001, 89, 416–421. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Zhou, X.-J.; Zhang, H. What Genetics Tells Us About the Pathogenesis of IgA Nephropathy: The Role of Immune Factors and Infection. Kidney Int. Rep. 2017, 2, 318–331. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, S.; Cui, W.; Gao, D.; Zhou, W.; Luo, P. Identification of potential biomarkers and therapeutic targets for human IgA nephropathy and hypertensive nephropathy by bioinformatics analysis. Mol. Med. Rep. 2017, 16, 3087–3094. [Google Scholar] [CrossRef]
- Tycová, I.; Hrubá, P.; Maixnerová, D.; Girmanová, E.; Mrázová, P.; Straňavová, L.; Zachoval, R.; Merta, M.; Slatinská, J.; Kollár, M.; et al. Molecular profiling in IgA nephropathy and focal and segmental glomerulosclerosis. Physiol. Res. 2018, 67, 93–105. [Google Scholar] [CrossRef]
- Wardle, E.N. B Lymphocyte Stimulator and Autoimmune Disease. Saudi J. Kidney Dis. Transplant. 2004, 15, 155. [Google Scholar]
- Li, W.; Peng, X.; Liu, Y.; Liu, H.; Liu, F.; He, L.; Liu, Y.; Zhang, F.; Guo, C.; Chen, G.; et al. TLR9 and BAFF: Their expression in patients with IgA nephropathy. Mol. Med. Rep. 2014, 10, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wu, W.; Wen, Y.; Li, X. Hydroxychloroquine alleviates persistent proteinuria in IgA nephropathy. Int. Urol. Nephrol. 2017, 49, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Chen, C.S.; Yiang, G.T.; Cheng, P.W.; Chen, Y.L.; Chiu, H.C.; Liu, K.H.; Lee, W.C.; Li, C.J. The Emerging Role of Pathogenesis of IgA Nephropathy. J. Clin. Med. 2018, 7, 225. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R. Treatment of IgA nephropathy: Recent advances and prospects. Nephrol. Ther. 2018, 14, S13–S21. [Google Scholar] [CrossRef]
- Yuling, H.; Ruijing, X.; Xiang, J.; Yanping, J.; Lang, C.; Li, L.; Dingping, Y.; Xinti, T.; Jingyi, L.; Zhiqing, T.; et al. CD19+CD5+ B Cells in Primary IgA Nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2130–2139. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhang, L.; Zhao, P.W.; Ma, L.; Li, C.; Zou, H.B.; Jiang, Y.F. Functional implications of regulatory B cells in human IgA nephropathy. Scand. J. Immunol. 2014, 79, 51–60. [Google Scholar] [CrossRef]
- Coppo, R.; Camilla, R.; Amore, A.; Peruzzi, L.; Daprà, V.; Loiacono, E.; Vatrano, S.; Rollino, C.; Sepe, V.; Rampino, T.; et al. Toll-like receptor 4 expression is increased in circulating mononuclear cells of patients with immunoglobulin A nephropathy. Clin. Exp. Immunol. 2010, 159, 73–81.