Intestinal fibrosis is one of the most threatening complications of Crohn’s disease. Endoscopic and surgical approaches are currently the only options available and there is an urgent need for targeted anti-fibrotic therapy. Several molecules investigated in preclinical studies, which are awaiting clinical trials in humans, have proven effective in CD stricturing phenotype and may be available in the near future as additional weapons in preventing or reversing intestinal fibrosis.
- antifibrotic therapy
- Crohn’s disease
- IBD
- intestinal fibrosis
1. Introduction
2. Overview of the Main Mechanisms of Intestinal Fibrosis in CD
As mentioned above, the abnormal inflammatory stimulus due to CD is associated with uncontrolled activation of mesenchymal cells, resulting in excessive ECM deposition [10]. In addition, an imbalance between matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs), appears to be associated with increased ECM deposition and subsequent tissue fibrosis [11]. These mechanisms, together with the thickening of the muscle layer due to hyperplasia and hypertrophy of the smooth muscle cells [12], determine the development of fibrostenotic strictures in CD.
Here we will briefly outline the main players underlying the process of intestinal fibrosis. The correlation between these factors is in most cases unknown and elusive. The fifigure below shows some of these cellular and molecular players and their interaction in the fibrogenic process leading to intestinal stricture formation.
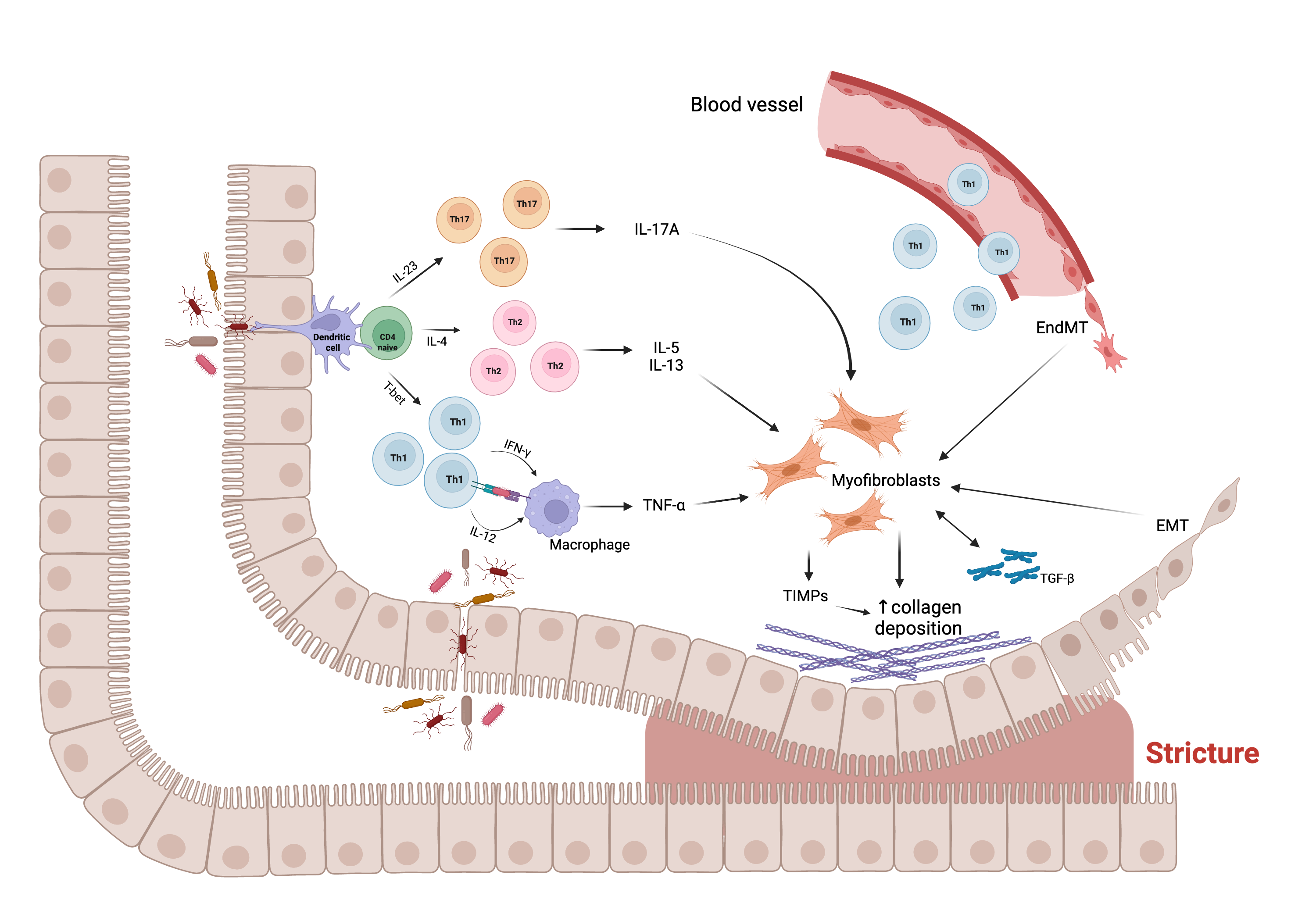
FigureFigure 1. Main molecular and cellular mechanisms, and their interaction, underlying the fibrogenic process leading to stricture formation in Crohn’s disease (CD). Intestinal mucosal infiltration of CD4+ T cells represents a key characteristic of CD. Multiple Th subsets have been identified, with different role in the fibrogenic process. The cross-talk between macrophage and T cells, sustained by Th1 pro-inflammatory cytokines, including interferon (IFN)-ɣ and interleukin (IL)-12, results in the production of tumor necrosis factor (TNF)-α, which promotes myofibroblast production of transforming growth factor (TGF)-β1. The latter inhibits the production of matrix metalloproteinases (MMPs) and favors the production of tissue inhibitor of metalloproteinases (TIMPs), which causes abnormal collagen deposition, with consequent fibrosis and stricture formation. In addition, Th2 and Th17 cells have a pro-fibrotic role through the production of pro- fibrotic cytokines, especially IL-17A, which induces intestinal myofibroblast secretion of collagen and TIMPs and significantly inhibits myofibroblast migration. The fibrotic process is also sustained by epithelial-mesenchymal transition (EMT) and endothelial-mesenchymal transition (EndMT), constantly evolving processes in which epithelial and endothelial cells acquire fibroblast characteristics. Abbreviations: EMT, epithelial-mesenchymal transition; EndMT, endothelial- mesenchymal transition; IL, interleukin; IFN, interferon; T-bet, T-box transcription factor; TGF, transforming growth factor; Th, T helper cell; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor; ↑, increase. Created with “BioRender.com”, 21 Jan 2022.
2.1. Main Cells Involved in Fibrogenesis
The main cells involved in the fibrogenesis process are mesenchymal cells, which are specifically committed to the production of collagen. The main players in this process are fibroblasts, myofibroblasts, and smooth muscle cells [13]. In particular, the inflammatory stimulus associated with CD seems to determine the activation of tissue fibroblasts and the migration of non-resident fibroblasts at the site of damage. These fibroblasts, under the stimulus of growth factors such as transforming growth factor (TGF)-β, may differentiate into myofibroblasts, capable of producing ECM. Similarly, smooth muscle cells are able to differentiate into myofibroblasts and likewise myofibroblasts can differentiate into smooth muscle cells and lead to the thickening of the muscularis propria and the formation of strictures [14]. Finally, the possible role of inflammation-induced differentiation of epithelial and endothelial cells into ECM-secreting mesenchymal cells should be considered, according to the mechanisms of epithelial-mesenchymal transition (EMT) and endothelial-mesenchymal transition (EndMT) [15,16][15][16]. EMT is a constantly evolving process in which epithelial cells acquire a migratory function and develop fibroblast characteristics. Similarly, EndMT is a process in which endothelial cells acquire fibroblast characteristics.
2.2. Molecular Mediators of Fibrosis
It is assumed that several cytokines can actively participate into the fibrogenesis process. Among these, the role of TGF-β is certainly predominant [17]. More specifically, the TGF-β1 isoform promotes collagen synthesis and fibroblast contraction in the mucosa of patients with fibrostenosing CD, acting through the Smad2-Smad3 molecular pathway and the regulation of TIMPs. Other cytokines related to organ fibrosis and with an emerging role in intestinal fibrosis, besides their known pro-inflammatory properties, are those belonging to the interleukin (IL)-1 family, including IL-1, IL-33, and IL-36 [18–21]. CD4+ T cells play a crucial role in the pathogenesis of CD and several T helper (Th) subsets have been identified, with different roles. While T-regulatory cells prevail in normal conditions, the Th1 subset appears predominantly pro-inflammatory, whereas Th2 and Th17 subsets appear to have both pro-inflammatory and pro-fibrogenic roles [22,23]. In particular, Th17 cells produce both IL-17 and IL-22 with a possible contrasting effect on intestinal fibrogenesis [24]. The role of cytokines belonging to the IL-17 family is well established, especially that of the IL-17A, as it induces intestinal myofibroblast secretion of collagen and TIMPs and significantly inhibits myofibroblast migration [25]. A possible role in this process has also been ascribed to the IL-17E (also known as IL-25), whose production in the human gut is reduced by tumor necrosis factor (TNF)-α and enhanced by TGF-β1 [26]. However, the pro-fibrotic role of IL-17E in CD has been questioned by the finding of no-significant difference on IL-17E levels in strictured compared to non- strictured CD tissues [25]. Fibroblast activation protein (FAP) is another protein typically It is assumed that several cytokines can actively participate into the fibrogenesis process. Among these, the role of TGF-β is certainly predominant [17]. More specifically, the TGF-β1 isoform promotes collagen synthesis and fibroblast contraction in the mucosa of patients with fibrostenosing CD, acting through the Smad2-Smad3 molecular pathway and the regulation of TIMPs. Other cytokines related to organ fibrosis and with an emerging role in intestinal fibrosis, besides their known pro-inflammatory properties, are those belonging to the interleukin (IL)-1 family, including IL-1, IL-33, and IL-36 [18][19][20][21]. CD4+ T cells play a crucial role in the pathogenesis of CD and several T helper (Th) subsets have been identified, with different roles. While T-regulatory cells prevail in normal conditions, the Th1 subset appears predominantly pro-inflammatory, whereas Th2 and Th17 subsets appear to have both pro-inflammatory and pro-fibrogenic roles [22][23]. In particular, Th17 cells produce both IL-17 and IL-22 with a possible contrasting effect on intestinal fibrogenesis [24]. The role of cytokines belonging to the IL-17 family is well established, especially that of the IL-17A, as it induces intestinal myofibroblast secretion of collagen and TIMPs and significantly inhibits myofibroblast migration [25]. A possible role in this process has also been ascribed to the IL-17E (also known as IL-25), whose production in the human gut is reduced by tumor necrosis factor (TNF)-α and enhanced by TGF-β1 [26]. However, the pro-fibrotic role of IL-17E in CD has been questioned by the finding of no-significant difference on IL-17E levels in strictured compared to non-strictured CD tissues [25]. Fibroblast activation protein (FAP) is another protein typically produced by activated fibroblasts during wound healing and implicated in the fibrotic evolution of tissue damage [27]. FAP has been shown to be highly overexpressed in the submucosa and the muscle layer of stenotic CD, compared to non-stenotic CD [28]. In addition, other growth factors have an established role in gut fibrosis, especially the basic fibroblast growth factor (bFGF), which is overexpressed in patients with stricturing CD phenotype [29]. Concerning the role of TNF-α family members, there is growing evidence about the TNF-like cytokine 1A (TL1A), secreted from immune cells and binding the death domain receptor 3 (DR3) expressed on intestinal myofibroblasts [30]. TL1A is highly expressed in the fibrotic tissue of CD patients and a gene variant of the TL1A gene is associated with a higher risk of fibrotic strictures [31]. Finally, a possible role of neutrophil extracellular traps (NETs) has recently emerged. NETs are large, extracellular, web-like structures extruded by neutrophils under various conditions, especially immune response towards pathogens, representing a defense mechanism that, if dysregulated, can contribute to the pathogenesis of immune-related disorders [32]. NETs have been shown to mediate the in vitro activation of fibroblasts into myofibroblasts in fibrotic interstitial lung disease [33], and it has been suggested that this role may also be played in the gut [34].
2.3. MicroRNAs
MicroRNAs (miRNAs) are small non-coding ribonucleic acid (RNA) sequences that interfere with mRNA, causing, in most cases, an inhibition of translation [35]. The role of miRNAs on intestinal fibrosis in CD is relatively poorly established. Two families of miRNAs, miRNA-29 and miRNA-200, appear to be involved in this process. Specifically, miRNA-29a, -29b, and -29c were found to be down-regulated in CD strictured mucosa, with a role for miRNA-29b in modulating in vitro the expression of collagen I and III [36]. The miRNA 200 family appears to play a protective role against the development of EMT [37,38].
2.4. The Role of Gut Microbiota
The human gut hosts a complex and abundant aggregation of microbes, collectively referred to as the gut microbiota, whose compositional and metabolic alterations, defined as dysbiosis, have a pivotal role in IBD pathogenesis [39]. All intestinal immune and non- immune cell types express the pathogen recognition receptors, such us Toll-like receptors (TLRs) and nucleotide-binding and oligomerization domain (NOD)-like receptors, which provide the ability to respond to pathogen-associated molecular patterns [40]. It has been reported that in primary human intestinal fibroblasts, the TLR5 ligand flagellin (present in all flagellated bacteria) induces a pro-inflammatory and pro-fibrotic phenotype [41]. More recently, it was confirmed that flagellin derived from adherent-invasive Escherichia coli (AIEC), a micro-organism frequently isolated in the ileal tissue of CD, could bind the TLR5 expressed in intestinal epithelium, determining the expression of the IL-33 receptor (ST2), which is crucial for the development of intestinal fibrosis [42]. Supporting the hypothesis of a causal role of microbiota on intestinal fibrogenesis, the recurrence of NOD2 variants in CD patients with fibrostenotic phenotype has been highlighted [43]. Finally, several studies on animal models of intestinal fibrosis have shown a pro-fibrotic activity of the gut microbiota [44,45].
2.4. The Role of Gut Microbiota
2.5. Matrix Stiffness
2.5. Matrix Stiffness
Another novel mechanism associated with intestinal fibrosis in CD patients is matrix stiffness. Resistance to matrix deformation has been shown to be an important mediator of cellular behavior [46]. Cell proliferation and differentiation are assumed to increase with matrix stiffness. This is true even for human colonic fibroblasts, which have been proved to be activated to a profibrotic phenotype by matrix stiffness [47,48]. An increase in ECM stiffness would be associated with a morphological alteration of fibroblasts, actin stress fiber formation and focal adhesion, promoting fibroblast proliferation and activation, even in the absence of inflammatory stimulation. This finding suggests that intestinal fibrosis would have a self-propagation mechanism independent of the inflammatory stimulus and matrix stiffness may have a role in this process.
2.6. “Creeping Fat”
2.6.“Creeping Fat”
Mesenteric fat and its hypertrophy, known as ‘creeping fat’, have an emerging role in the pathogenesis of CD fibrostenotic phenotype [49]. The role of creeping fat in intestinal fibrogenesis is associated with the production of adipokines that promote a shift of the macrophage compartment towards M2 macrophage dominance, resulting in increased production of TGF-β1 [50]. Furthermore, creeping fat has been shown to be associated with smooth muscle hypertrophy [51].
3. Therapeutic Approaches of Crohn’s Disease
To date, there are no medical therapies available to prevent or reverse intestinal fibrosis in CD. In patients with intestinal obstruction due to CD with a fibrostenosing phenotype, initial treatment consists of nasogastric decompression, bowel rest, intravenous hydration, and electrolyte replacement. Subsequent management is dependent on strictures’ inflammation degree and morphometrics, such as location and length, assessed by biomarkers (e.g., C-reactive protein, erythrocyte sedimentation rate, and fecal calprotectin), endoscopy, computed tomography, or magnetic resonance imaging. Current complications (such as phlegmon, abscess, dysplasia, or malignancy) and patient preferences should also be taken into account [5,54][5][52].
3.1. Current Medical Options
3.2. Endoscopic and Surgical Management
3.3. Promising Anti-Fibrotic Therapy in CD
Increasing knowledge of the molecular mechanisms underlying intestinal fibrosis has enabled the identification of anti-fibrotic therapeutic targets. At present, although there is no therapy capable of treating or reversing intestinal fibrosis in CD, several pre- clinical studies have been conducted in vivo, ex vivo, and in vitro, with encouraging results. Herein, we report the most promising anti-fibrotic therapeutic targets known to date and the relevant target-specific molecules under investigation (Table below)are summarized in Table 1.
3.3.1. Targeting TGF-β Pathways
The most promising target for anti-fibrotic therapy is TGF-β, the principal molecular mediator of fibrogenesis, and its signaling pathways.
TNBS-colitis | ||||
90 | ||||
|
Pirfenidone |
Smad, PI3K/AKT, MAPK, and mTOR signaling pathways inhibition |
HIFs, DSS-colitis, RIF |
93-96 |
|
|
Maggot extract |
↑ Nrf2 expression |
DSS-colitis |
98 |
|
|
TIMP/MMP balance |
Thalidomide |
Altered TIMP/MMPs balance and ECM degradation |
TNBS-colitis |
100 |
|
VEGF |
Bevacizumab |
↓ collagen deposition |
n.a. |
n.a. |
|
FAP |
Anti-FAP Ab |
FAP inhibition |
HIFs |
104 |
|
EMT |
rhBMP-7 |
EMT inhibition |
TNBS-colitis |
111 |
|
miRNA200b-containing microvescicles |
EMT inhibition |
TNBS-colitis, IEC-6 |
112 |
|
|
Endogenous cannabinoid system |
MAEA |
↓ collagen production and ↑ myofibroblasts migration |
Human organ culture biopsies, LPMCs, and HIFs |
117 |
|
IL-17 |
Anti-IL17 Ab |
↓ profibrogenic cytokines and MMP/TIMPs balance alteration |
TNBS-colitis |
119 |
|
IL-36 |
Anti-IL36R Ab |
↓ collagen production, MMPs, IL6 signaling, and EMT |
DSS- and TNBS-colitis |
124 |
|
TL1A |
Anti-TL1A Ab |
TGF-1/Smad3 signaling pathway inhibition |
T-cell transfer-colitis |
125 |
|
TNF-α and IL-17 |
ABT-122 |
n.a. |
n.a. |
n.a. |
|
AXL pathway |
BGB324 |
↓ matrix stiffness and TGF-b1-induced fibrogenesis |
CCD-18co, TNBS-colitis |
131 |
|
NETs |
PAD4 inhibitors |
↓ NETs-derived fibrosis |
n.a. |
n.a. |
|
miRNA |
miRNA29 |
↓ TGF-β1-induced collagen expression |
Human fibroblasts cultures |
36 |
|
miRNA200 |
↓ ZEB1 and ZEB2, EMT inhibition |
Intestinal epithelial cells |
38 |
|
|
Matrix Stiffness |
b-aminopropionitrile |
↑ MMP3 activity and ↓ ECM contraction |
HIFs |
137 |
|
Gut microbiota |
-
Several studies on fibrosis of other tissues have shown that TGF-β1 production was strongly stimulated by the local activation of angiotensin II [72,73,74][70][71][72], the main effector of the renin-angiotensin system, whose activity is increased in the colonic mucosa of CD patients [75][73]. For this reason, it was assumed that angiotensin conversing enzyme (ACE) inhibitors and sartans (angiotensin II receptor antagonists), which typically act as anti-hypertensives, could also play a role in the process of intestinal fibrogenesis. The first ACE-inhibitor investigated was captopril, which showed to be effective in preventing colonic fibrosis in 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis in rats. Its anti-fibrotic action has been assumed to derive from blocking TGF-β1 overexpression and/or from a direct down-regulation of TGF-β1 transcripts [76][74]. Moreover, transanal administration of enalaprilat has been shown to be effective in preventing colonic fibrosis in a dextran sulfate sodium (DSS)-induced colitis model [77][75]. More recently, losartan, an antagonist of the angiotensin II receptor, was investigated and exhibited a pleiotropic effect, reducing TGF-β1 concentration and significantly improving the macro- and microscopic scores of fibrosis in the colonic wall of rats [78][76];
-
Based on the known antagonistic relationship between the TGF-β/Smad pathway and the peroxisome proliferator-activated receptor (PPAR)γ, a member of ligand-activated transcription factors of nuclear hormone receptor superfamily [79,80][77][78], the effect of a novel 5-ASA analog (named GED-0507-34 Levo), able to activate PPARγ, has been investigated. GED-0507-34 Levo showed improvement of intestinal fibrosis in DSS-induced chronic colitis in mice, reducing the activation of myofibroblasts and the expression of the main pro-fibrotic molecules including TGF-β, Smad3, IL-13 and connective tissue growth factor (CTGF) [81][79]. Similarly, it has been shown that other PPARγ agonists, usually employed in the treatment of diabetes, such as troglitazone and rosiglitazone, may be useful in counteracting the fibrogenic process by suppressing TGF-β1-induced synthesis of collagen, fibronectin, and α-smooth muscle actin in human primary intestinal myofibroblasts [82][80];
-
Another target signaling pathway induced by TGF-β1 but also by matrix stiffness is that of Rho/Rho chinase (ROCK) [83][81]. The first ROCK inhibitors studied were CCG-1423, CCG-100602, and CCG-203971, which, by inhibiting RhoA signaling in myofibroblasts, induced a significant anti-fibrotic activity [84,85][82][83]. These molecules, however, showed an unacceptable toxicity profile, especially with regard to cardiovascular side effects [86][84]. For this reason, the effect of a locally acting ROCK inhibitor (AMA0825) was investigated. This molecule prevented and reversed intestinal fibrosis in vitro and ex vivo by diminishing TGF-β1-induced activation of myocardine-related transcription factor and p38 mitogen-activated protein kinase (MAPK) and increasing autophagy in fibroblasts, with a good tolerability profile [87][85]. Combining AMA0825 with anti-inflammatory agents (such as anti-TNF-α) in vivo ameliorated inflammation but also prevented accumulation of fibrotic tissue, underscoring the importance of combination therapy;
-
Other compounds have been shown to downregulate the TGF-β signaling. These include cilengitide, which is an Arg-Gly-Asp (RGD)-containing αVβ3 integrin inhibitor, that is able to decrease TGF-β1 activation and development of fibrosis in chronic TNBS-induced colitis [88][86]. More recently, anti-fibrotic intestinal efficacy has been proposed for two molecules approved for the treatment of idiopathic pulmonary fibrosis, namely pirfenidone and nintedanib [89,90][87][88]. In particular, pirfenidone, an orally delivered pyridine derivative that suppresses TGF-β and TNF-α signals, inhibited, both in vivo and in vitro, intestinal fibroblast proliferation and motility and reduced collagen production through different TGF-β1 signaling pathways, including those of suppressor of mothers against decapentaplegic (Smad), phosphatidylinositol-3-kinase (PI3K)/AKT, MAPK, and mechanistic target of rapamycin (mTOR) [91,92,93,94][89][90][91][92]. Therefore, this molecule is of great interest and has important therapeutic potential, but needs further studies to better clarify its mechanism of action, efficacy, and safety [95][93]. No studies are yet available on the usefulness in intestinal fibrosis of nintedanib, a small oral molecule inhibitor of tyrosine kinase receptors, such as platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF) receptors. Finally, an anti-fibrotic action of maggot extract was described by downregulating the TGF-β1/Smad pathway via upregulation of nuclear factor erythroid 2-related factor 2 (Nrf2) expression [96][94].
3.3.2. Targeting TIMP/MMP Balance
3.3.3. Targeting VEGF
3.3.4. Targeting FAP
3.3.5. Targeting EMT
3.3.6. Targeting the Endogenous Cannabinoid System
3.3.7. Targeting IL-17
3.3.8. Targeting IL-36
3.3.9. Targeting TL1A
3.3.10. Targeting Both TNF-α and IL-17
3.3.11. Targeting AXL Pathway
3.3.12. Targeting NETs
3.3.13. Targeting miRNAs
3.3.14. Targeting Matrix Stiffness
3.3.15. Targeting Intestinal Microbiota
|
TARGET |
AGENT |
MECHANISM |
MODEL |
REFERENCE |
|
TGF-b pathways |
Captopril |
↓ TGF-b1 expression and/or TGF-b1 transcript |
TNBS-colitis |
78 |
|
Transanal enalaprilat |
↓ TGF-b signaling pathway |
DSS-colitis |
79 |
|
|
Losartan |
↓ TGF-b1 expression |
TNBS-colitis |
80 |
|
|
GED-0507-34 Levo |
PPAR-γ activation |
DSS-colitis |
83 |
|
|
Troglitazone, Rosiglitazone |
PPAR-γ activation |
HIFs |
84 |
|
|
CCG-1423, CCG-100602, CCG-203971 |
ROCK inhibition |
CCD18-co HIFs |
87 |
|
|
AMA0825 |
ROCK inhibition |
DSS- and T-cell transfer-colitis, HIFs |
89 |
|
|
Cilengitide |
αVβ3 integrin inhibition | |||
Probiotics and prebiotics | ||||
Modulation fibrotic pathways | ||||
Mouse and cellular models | ||||
138-142 | ||||
Abbreviations: Ab, antibody; CCD-18Co, noncancerous colon fibroblast; DSS, dextran sulfate sodium; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; EndMT, endothelial mesenchymal transition; FAP, fibroblast activation protein; HIF, human intestinal fibroblast; IEC, intestinal epithelial cell; IL, interleukin; LPMC, lamina propria mononuclear cell; MAEA, methanandamide; MAPK, mitogen-activated protein kinase; mTOR, mechanistic target of rapamycin; miRNA, micro ribonucleic acid; MMP, matrix metalloproteinase; n.a., not available; NET, neutrophil extracellular trap; Nrf2, nuclear factor erythroid 2-related factor 2; PAD4, peptidylarginine deiminase 4; PI3K, phosphatidylinositol-3-Kinase; PPAR, peroxisome proliferator-activated receptor; rhBMP-7, recombinant human bone morphogenic protein-7; RIF, radiation-induced intestinal fibrosis; ROCK, Rho/Rho chinase; Smad, suppressor of mothers against decapentaplegic; TGF, transforming growth factor; TIMP, tissue inhibitor of metalloproteinase; TL1A, TNF-like cytokine 1A; TNBS, 2,4,6-trinitrobenzene sulfonic acid; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor; ZEB, zinc finger E-box binding homeobox; ↑, increase; ↓, decrease.
4. Major Challenge for Anti-Fibrotic Agents Development
There are numerous pitfalls in identifying anti-fibrotic drugs for CD. First, experimental fibrosis in cellular and animal models does not necessarily resemble human fibrosis [139]. Cells may behave differently in vitro and in vivo and single cell studies often do not reproduce the complex in vivo cellular network. For this reason, 3D models are under development to reproduce the natural microenvironment as closely as possible to that in vivo [140,141]. Moreover, the targeted molecules often represent a small component of the complex molecular maze underlying the fibrotic process. The target molecules may even have multiple functions on the intestinal tissue, with the risk of targeting processes implicated in physiological tissue remodeling, resulting in negative effects. In addition, to date there are neither fibrosis biomarkers, nor diagnostic tools that can be used to identify and quantify the overall fibrotic burden in CD patients, especially in the early stages, when anti-fibrotic therapy may be mostly effective [142,143]. Finally, there is an urgent need of end points that can be used to assess the efficacy of anti-fibrotic agents in clinical trials. For this reason, several groups of renowned IBD experts have reached expert consensus on this matter [6,144]. In particular, a core set of 13 end-points (i.e., complete clinical response, long-term efficacy, sustained clinical benefit, treatment failure, radiological remission, normal quality of life, clinical remission without steroids, therapeutic failure, deep remission, complete absence of occlusive symptoms, symptom- free survival, bowel damage progression, and no disability) were considered critical [144]. The combination of improved clinical, endoscopic and/or radiological features seems appropriate to define a successful treatment [6]. The need for intervention within 24-48 weeks from medical therapy has been proposed as the most accurate end-point to assess anti-fibrotic agents in pharmacological trials.
Despite the urgency for anti-fibrotic therapy and the numerous molecules identified as potential anti-fibrotics in CD, no phase III clinical trial is currently ongoing or recruiting to our knowledge (according to ClinicalTrials.gov, as of 19 January 2022).
References
- Baumgart, D.C.; Sandborn, W.J. Chron’s disease. Lancet 2012, 380, 1590–1605.
- Kaplan, G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727.
- Di Sabatino, A.; Rovedatti, L. Recent advances in understanding Crohn’s disease. Intern. Emerg. Med. 2013, 8, 101–113.
- Knowles, S.R.; Graff, L.A. Quality of life in inflammatory bowel disease: A systematic review and meta-analyses-Part I. Inflamm. Bowel Dis. 2018, 24, 742–751.
- Rieder, F.; Fiocchi, C. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology 2017, 152, 340–350.
- Rieder, F.; Bettenworth, D. An expert consensus to standardise definitions, diagnosis and treatment targets for anti-fibrotic stricture therapies in Crohn’s disease. Aliment Pharmacol. Ther. 2018, 48, 347–357.
- Shaw, T.J.; Martin, P. Wound repair at a glance. J. Cell Sci. 2009, 122, 3209–3213.
- Xue, M.; Jackson, C.J. Extracellular Matrix Reorganization During Wound Healing and Its Impact on Abnormal Scarring. Adv. Wound Care 2015, 4, 119–136.
- Pakshir, P.; Boris, H. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68-69, 81–93.
- Lenti, M.V.; Di Sabatino, A. Intestinal fibrosis. Mol. Aspects Med. 2019, 65, 100–109.
- Medina, C.; Santos-Martinez, M.J. Transforming growth factor-beta type 1 receptor (ALK5) and Smad proteins mediate TIMP-1 and collagen synthesis in experimental intestinal fibrosis. J. Pathol. 2011, 224, 461–472.
- Chen, W.; Lu, C. Smooth muscle hyperplasia/hypertrophy is the most prominent histological change in Crohn’s fibrostenosing bowel strictures: A semiquantitative analysis by using a novel histological grading scheme. J. Crohn’s Colitis 2017, 11, 92–104.
- Rieder, F.; Fiocchi, C. Intestinal fibrosis in IBD-a dynamic, multifactorial process. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 228–235.
- Speca, S.; Giusti, I. Cellular and molecular mechanisms of intestinal fibrosis. World J. Gastroenterol. 2012, 18, 3635–3661.
- Jiang, H.; Shen, J. Epithelial-mesenchymal transition in Crohn’s disease. Mucosal Immunol. 2018, 11, 294–303.
- Rieder, F.; Kessler, S.P. Inflammation-induced endothelial-to-mesenchymal transition: A novel mechanism of intestinal fibrosis. Am. J. Pathol. 2011, 179, 2660–2673.
- Di Sabatino, A.; Jackson, C.L. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut 2009, 58, 777–789.
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27.
- Drygiannakis, I.; Valatas, V. Proinflammatory cytokines induce crosstalk between colonic epithelial cells and subepithelial myofibroblasts: Implication in intestinal fibrosis. J. Crohn’s Colitis 2013, 7, 286–300.
- Lopetuso, L.R.; Scaldaferri, F. Emerging role of the interleukin (IL)-33/ST2 axis in gut mucosal wound healing and fibrosis. Fibrogenesis Tissue Repair. 2012, 5, 18.
- Elias, M.; Zhao, S. IL-36 in chronic inflammation and fibrosis—bridging the gap? J. Clin. Investig. 2021, 131, e144336.
- Tindemans, I.; Joose, M.E. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110.
- Friedrich, M.; Pohin, M. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006.
- Latella, G.; Viscido, A. Controversial Contribution of Th17/IL-17 Toward the Immune Response in Intestinal Fibrosis. Dig. Dis. Sci. 2020, 65, 1299–1306.
- Biancheri, P.; Pender, S.L. The role of interleukin 17 in Crohn’s disease-associated intestinal fibrosis. Fibrogenesis Tissue Repair. 2013, 6, 13.
- Fina, D.; Franzè, E. Interleukin-25 production is differently regulated by TNF-α and TGF-β1 in the human gut. Mucosal. Immunol. 2011, 4, 239–244.
- Rovedatti, L.; Di Sabatino, A. Fibroblast activation protein expression in Crohn’s disease strictures. Inflamm. Bowel Dis. 2011, 17, 1251–1253.
- Truffi, M.; Sorrentino, L. Inhibition of Fibroblast Activation Protein Restores a Balanced Extracellular Matrix and Reduces Fibrosis in Crohn’s Disease Strictures Ex Vivo. Inflamm. Bowel Dis. 2018, 24, 332–345.
- Di Sabatino, A.; Ciccocioppo, R. Serum bFGF and VEGF correlate respectively with bowel wall thickness and intramural blood flow in Crohn’s disease. Inflamm. Bowel Dis. 2004, 10, 573–577.
- Shih, D.Q.; Zheng, L. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol. 2014, 7, 1492–1503.
- Barrett, R.; Zhang, X. Constitutive TL1A expression under colitogenic conditions modulates the severity and location of gut mucosal inflammation and induces fibrostenosis. Am. J. Pathol. 2012, 180, 636–649.
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147.
- Chrysanthopoulou, A.; Mitroulis, I. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307.
- Dragoni, G.; De Hertogh, G. The Role of Citrullination in Inflammatory Bowel Disease: A Neglected Player in Triggering Inflammation and Fibrosis? Inflamm. Bowel Dis. 2021, 27, 134–144.
- Voglova, K.; Bezakova, J. Micro RNAs: An arguable appraisal in medicine. Endocr. Regul. 2016, 50, 106–124.
- Nijhuis, A.; Biancheri, P. In Crohn’s disease fibrosis-reduced expression of the miR-29 family enhances collagen expression in intestinal fibroblasts. Clin. Sci. 2014, 127, 341–350.
- Mongroo, P.S.; Rustgi, A.K. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol. Ther. 2010, 10, 219–222.
- Chen, Y.; Ge, W. miR-200b is involved in intestinal fibrosis of Crohn’s disease. Int. J. Mol. Med. 2012, 29, 601–606.
- Nishida, A.; Inoue, R. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10.
- Rieder, F. The gut microbiome in intestinal fibrosis: Environmental protector or provocateur? Sci. Transl. Med. 2013, 5, 1–90.
- Rieder, F.; Bhilocha, S. Activation of toll-like receptor (TLR) 5 induces a pro-fibrogenic phenotype on human intestinal myofibroblasts (HIF)—A novel pathway mediated by Caspase 1. Gastroenterology 2011, 140, S114.
- Imai, J.; Kitamoto, S. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019, 12, 632–643.
- Abreu, M.T.; Taylor, K.D. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology 2002, 123, 679–688.
- Grassl, G.A.; Valdez, Y. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology 2008, 134, 768–780.
- Mourelle, M.; Salas, A. Stimulation of transforming growth factor beta1 by enteric bacteria in the pathogenesis of rat intestinal fibrosis. Gastroenterology 1998, 114, 519–526.
- Wells, R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 2008, 47, 1394–1400.
- Johnson, L.A.; Rodansky, E.S. Matrix stiffness corresponding to strictured bowel induces a fibrogenic response in human colonic fibroblasts. Inflamm. Bowel Dis. 2013, 19, 891–903.
- Johnson, L.A.; Luke, A. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: Impact of a "Top-Down" approach to intestinal fibrosis in mice. Inflamm. Bowel Dis. 2012, 18, 460–471.
- Peyrin-Biroulet, L.; Chamaillard, M. Mesenteric fat in Crohn’s disease: A pathogenetic hallmark or an innocent bystander? Gut 2007, 56, 577–583.
- Kredel, L.I.; Batra, A. Adipokines from local fat cells shape the macrophage compartment of the creeping fat in Crohn’s disease. Gut 2013, 62, 852–862.
- Mao, R.; Kurada, S. The Mesenteric Fat and Intestinal Muscle Interface: Creeping Fat Influencing Stricture Formation in Crohn’s Disease. Inflamm. Bowel Dis. 2019, 25, 421–426.
- Lu, C.; Baraty, B. Systematic review: Medical therapy for fibrostenosing Crohn’s disease. Aliment Pharmacol. Ther. 2020, 51, 1233–1246.
- Yaffe, B.H.; Korelitz, B.I. Prognosis for nonoperative management of small-bowel obstruction in Crohn’s disease. J. Clin. Gastroenterol. 1983, 5, 211–215.
- Vasilopoulos, S.; Kugathasan, S. Intestinal strictures complicating initially successful infliximab treatment for luminal Crohn’s disease. Am. J. Gastroenterol. 2000, 95, 2503.
- Toy, L.S.; Scherl, E.J. Complete bowel obstruction following initial response to infliximab therapy for Crohn’s disease: A series of a newly described complication. Gastroenterology 2000, 118, A569.
- Allocca, M.; Bonifacio, C. Efficacy of tumour necrosis factor antagonists in stricturing Crohn’s disease: A tertiary center real-life experience. Dig. Liver Dis. 2017, 49, 872–877.
- Hanauer, S.B.; Feagan, B.G. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002, 359, 1541–1549.
- Bouhnik, Y.; Carbonnel, F. Efficacy of adalimumab in patients with Crohn’s disease and symptomatic small bowel stricture: A multicentre, prospective, observational cohort (CREOLE) study. Gut 2018, 67, 53–60.
- Engel, T.; Ungar, B. Vedolizumab in IBD-Lessons from Real-world Experience; A Systematic Review and Pooled Analysis. J. Crohn’s Colitis 2018, 12, 245–257.
- Ma, C.; Fedorak, R.N. Clinical, endoscopic and radiographic outcomes with ustekinumab in medically-refractory Crohn’s disease: Real world experience from a multicentre cohort. Aliment Pharmacol. Ther. 2017, 45, 1232–1243.
- Gordon, M. 5-Aminosalicylates to maintain remission in Crohn’s disease: Interpreting conflicting systematic review evidence. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 99–102.
- Rieder, F.; Latella, G. European Crohn’s and Colitis Organisation Topical Review on Prediction, Diagnosis and Management of Fibrostenosing Crohn’s Disease. J. Crohns Colitis 2016, 10, 873–885.
- Hirai, F.; Andoh, A. Efficacy of Endoscopic Balloon Dilation for Small Bowel Strictures in Patients With Crohn’s Disease: A Nationwide, Multi-centre, Open-label, Prospective Cohort Study. J. Crohns Colitis 2018, 12, 394–401.
- Bettenworth, D.; Gustavsson, A. A Pooled Analysis of Efficacy, Safety, and Long-term Outcome of Endoscopic Balloon Dilation Therapy for Patients with Stricturing Crohn’s Disease. Inflamm. Bowel Dis. 2017, 23, 133–142.
- Siddiqui, U.D.; Banerjee, S. Tools for endoscopic stricture dilation. Gastrointest. Endosc. 2013, 78, 391–404.
- Bemelman, W.A.; Warusavitarne, J. ECCO-ESCP Consensus on Surgery for Crohn’s Disease. J. Crohns Colitis 2018, 12, 1–16.
- Katsuno, H.; Maeda, K. Novel antimesenteric functional end-to-end handsewn (Kono-S) anastomoses for Crohn’s disease: A report of surgical procedure and short-term outcomes. Dig. Surg. 2015, 32, 39–44.
- Coffey, C.J.; Kiernan, M.G. Inclusion of the Mesentery in Ileocolic Resection for Crohn’s Disease is Associated with Reduced Surgical Recurrence. J. Crohns Colitis 2018, 12, 1139–1150.
- Sica, G.S.; Iaculli, E. Laparoscopic versus open ileo-colonic resection in Crohn’s disease: Short-and long-term results from a prospective longitudinal study. J. Gastrointest. Surg. 2008, 12, 1094–1102.
- Campbell, S.E.; Katwa, L.C. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J. Mol. Cell Cardiol. 1997, 29, 1947–1958.
- Warner, F.J.; Lubel, J.S. Liver fibrosis: A balance of ACEs? Clin. Sci. 2007, 113, 109–118.
- Uhal, B.D.; Kim, J.K. Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary fibrosis: Autocrine mechanisms in myofibroblasts and macrophages. Curr. Pharm. Des. 2007, 13, 1247–1256.
- Jaszewski, R.; Tolia, V. Increased colonic mucosal angiotensin I and II concentrations in Crohn’s colitis. Gastroenterology 1990, 98, 1543–1548.
- Wengrower, D.; Zanninelli, G. Prevention of fibrosis in experimental colitis by captopril: The role of tgf-beta1. Inflamm. Bowel Dis. 2004, 10, 536–545.
- Koga, H.; Yang, H. Transanal delivery of angiotensin converting enzyme inhibitor prevents colonic fibrosis in a mouse colitis model: Development of a unique mode of treatment. Surgery 2008, 144, 259–268.
- Wengrower, D.; Zanninelli, G. Losartan reduces trinitrobenzene sulphonic acid-induced colorectal fibrosis in rats. Can. J. Gastroenterol. 2012, 26, 33–39.
- Wei, J.; Ghosh, A.K. PPARγ downregulation by TGFß in fibroblast and impaired expression and function in systemic sclerosis: A novel mechanism for progressive fibrogenesis. PLoS ONE 2010, 5, e13778.
- Ghosh, A.K.; Bhattacharyya, S. Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J. 2009, 23, 2968–2977.
- Speca, S.; Rousseaux, C. Novel PPARγ Modulator GED-0507-34 Levo Ameliorates Inflammation-driven Intestinal Fibrosis. Inflamm. Bowel Dis. 2016, 22, 279–292.
- Koo, J.B.; Nam, M.O. Anti-fibrogenic effect of PPAR-γ agonists in human intestinal myofibroblasts. BMC Gastroenterol. 2017, 17, 73.
- Bourgier, C.; Haydont, V. Inhibition of Rho kinase modulates radiation induced fibrogenic phenotype in intestinal smooth muscle cells through alteration of the cytoskeleton and connective tissue growth factor expression. Gut 2005, 54, 336–343.
- Sandbo, N.; Kregel, S. Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-beta. Am. J. Respir. Cell Mol. Biol. 2009, 41, 332–338.
- Johnson, L.A.; Rodansky, E.S. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-β-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel Dis. 2014, 20, 154–165.
- Bian, H.; Zhou, Y. Rho-kinase signaling pathway promotes the expression of PARP to accelerate cardiomyocyte apoptosis in ischemia/reperfusion. Mol. Med. Rep. 2017, 16, 2002–2008.
- Holvoet, T.; Devriese, S. Treatment of Intestinal Fibrosis in Experimental Inflammatory Bowel Disease by the Pleiotropic Actions of a Local Rho Kinase Inhibitor. Gastroenterology 2017, 153, 1054–1067.
- Li, C.; Flynn, R.S. Increased activation of latent TGF-β1 by αVβ3 in human Crohn’s disease and fibrosis in TNBS colitis can be prevented by cilengitide. Inflamm. Bowel Dis. 2013, 19, 2829–2839.
- Sathiyamoorthy, G.; Sehgal, S. Pirfenidone and Nintedanib for Treatment of Idiopathic Pulmonary Fibrosis. South Med. J. 2017, 110, 393–398.
- Ma, C.; Jairath, V. Targeting anti-fibrotic pathways in Crohn’s disease—The final frontier? Best Pract. Res. Clin. Gastroenterol. 2019, 38-39, 101603.
- Cui, Y.; Zhang, M. Pirfenidone Inhibits Cell Proliferation and Collagen I Production of Primary Human Intestinal Fibroblasts. Cells. 2020, 9, 775.
- Sun, Y.; Zhang, Y. Pirfenidone suppresses TGF-β1-induced human intestinal fibroblasts activities by regulating proliferation and apoptosis via the inhibition of the Smad and PI3K/AKT signaling pathway. Mol. Med. Rep. 2018, 18, 3907–3913.
- Li, G.; Ren, J. Oral pirfenidone protects against fibrosis by inhibiting fibroblast proliferation and TGF-β signaling in a murine colitis model. Biochem. Pharmacol. 2016, 117, 57–67.
- Sun, Y.W.; Zhang, Y.Y. Pirfenidone prevents radiation-induced intestinal fibrosis in rats by inhibiting fibroblast proliferation and differentiation and suppressing the TGF-β1/Smad/CTGF signaling pathway. Eur. J. Pharmacol. 2018, 822, 199–206.
- Latella, G.; Viscido, A. Could Pirfenidone Also be Effective in Treating Intestinal Fibrosis? Cells 2020, 9, 1762.
- Wang, R.; Wang, D. Therapeutic Targeting of Nrf2 Signaling by Maggot Extracts Ameliorates Inflammation-Associated Intestinal Fibrosis in Chronic DSS-Induced Colitis. Front. Immunol. 2021, 12, 670159.
- Zhu, Z.; Li, M.; Shu, X. Thalidomide is a therapeutic agent that is effective in inducing and maintaining endoscopic remission in adult CD patients. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 210–216.
- Chen, H.; Xu, H. Thalidomide Prevented and Ameliorated Pathogenesis of Crohn’s Disease in Mice via Regulation of Inflammatory Response and Fibrosis. Front. Pharmacol. 2019, 10, 1486.
- Chu, S.J.; Zhang, Z.H. Effect of bevacizumab on the expression of fibrosis-related inflammatory mediators in ARPE-19 cells. Int. J. Ophthalmol. 2017, 10, 366–371.
- Alkim, C.; Alkim, H. Angiogenesis in Inflammatory Bowel Disease. Int. J. Inflam. 2015, 2015, 970890.
- Juillerat-Jeanneret, L.; Gerber-Lemaire, S. The prolyl-aminodipeptidases and their inhibitors as therapeutic targets for fibrogenic disorders. Mini Rev. Med. Chem. 2009, 9, 215–226.
- Egger, C.; Cannet, C. Effects of the fibroblast activation protein inhibitor, PT100, in a murine model of pulmonary fibrosis. Eur. J. Pharmacol. 2017, 809, 64–72.
- Hill, C.; Li, J. Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis. 2019, 10, 591.
- Yao, L.; Conforti, F. Paracrine signalling during ZEB1-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957.
- Zeisberg, M.; Yang, C. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347.
- Zeisberg, E.M.; Tarnavski, O. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961.
- Zeisberg, M.; Hanai, J. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968.
- Flier, S.N.; Tanjore, H. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212.
- Yang, J.; Zhou, C.Z. miR-200b-containing microvesicles attenuate experimental colitis associated intestinal fibrosis by inhibiting epithelial-mesenchymal transition. J. Gastroenterol. Hepatol. 2017, 32, 1966–1974.
- Muccioli, G.G. Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discov. Today 2010, 15, 474–483.
- Petrosino, S.; Ligresti, A. Endocannabinoid chemical biology: A tool for the development of novel therapies. Curr. Opin. Chem. Biol. 2009, 13, 309–320.
- Izzo, A.A.; Camilleri, M. Emerging role of cannabinoids in gastrointestinal and liver diseases: Basic and clinical aspects. Gut 2008, 57, 1140–1155.
- Julien, B.; Grenard, P. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 2005, 128, 742–755.
- Di Sabatino, A.; Battista, N. The endogenous cannabinoid system in the gut of patients with inflammatory bowel disease. Mucosal Immunol. 2011, 4, 574–583.
- Li, J.; Liu, L. Role of Interleukin-17 in Pathogenesis of Intestinal Fibrosis in Mice. Dig. Dis. Sci. 2020, 65, 1971–1979.
- Paul, J.; Singh, A.K. IL-17-driven intestinal fibrosis is inhibited by Itch-mediated ubiquitination of HIC-5. Mucosal Immunol. 2018, 11, 427–436.
- Hueber, W.; Sands, B.E. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700.
- Targan, S.R.; Feagan, B. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607.
- Zhang, B.L.; Liang, T.S. Interleukin-17 as a Therapy Target in Intestinal Fibrosis. Dig. Dis. Sci. 2020, 65, 3054–3055.
- Melton, E.; Qiu, H. Interleukin-36 Cytokine/Receptor Signaling: A New Target for Tissue Fibrosis. Int. J. Mol. Sci. 2020, 21, 6458.
- Scheibe, K.; Kersten, C. Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice with Chronic Intestinal Inflammation. Gastroenterology 2019, 156, 1082–1097.
- Danese, S.; Klopocka, M. Anti-TL1A Antibody PF-06480605 Safety and Efficacy for Ulcerative Colitis: A Phase 2a Single-Arm Study. Clin. Gastroenterol. Hepatol. 2021, 19, 2324–2332.
- Fleischmann, R.M.; Wagner, F. Safety, Tolerability, and Pharmacodynamics of ABT-122, a Tumor Necrosis Factor- and Interleukin-17-Targeted Dual Variable Domain Immunoglobulin, in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 2283–2291.
- Mease, P.J.; Genovese, M.C. Phase II Study of ABT-122, a Tumor Necrosis Factor- and Interleukin-17A-Targeted Dual Variable Domain Immunoglobulin, in Patients with Psoriatic Arthritis with an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2018, 70, 1778–1789.
- Baker, K.F.; Isaacs, J.D. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann. Rheum. Dis. 2018, 77, 175–187.
- Bárcena, C.; Stefanovic, M. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678.
- Steiner, C.A.; Rodansky, E.S. AXL Is a Potential Target for the Treatment of Intestinal Fibrosis. Inflamm. Bowel Dis. 2021, 27, 303–316.
- Gottlieb, Y.; Elhasid, R. Neutrophil extracellular traps in pediatric inflammatory bowel disease. Pathol. Int. 2018, 68, 517–523.
- He, Z.; Si, Y. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thromb. Haemost. 2016, 115, 738–751.
- Angelidou, I.; Chrysanthopoulou, A. REDD1/Autophagy Pathway Is Associated with Neutrophil-Driven IL-1β Inflammatory Response in Active Ulcerative Colitis. J. Immunol. 2018, 200, 3950–3961.
- Dinallo, V.; Marafini, I. Neutrophil Extracellular Traps Sustain Inflammatory Signals in Ulcerative Colitis. J. Crohn’s Colitis 2019, 13, 772–784.
- Suzuki, M.; Ikari, J. PAD4 Deficiency Improves Bleomycin-induced Neutrophil Extracellular Traps and Fibrosis in Mouse Lung. Am. J. Respir. Cell Mol. Biol. 2020, 63, 806–818.
- de Bruyn, J.R.; van den Brink, G.R. Fibrostenotic Phenotype of Myofibroblasts in Crohn’s Disease is Dependent on Tissue Stiffness and Reversed by LOX Inhibition. J. Crohn’s Colitis 2018, 12, 849–859.
- Kashima, S.; Fujiya, M. Polyphosphate, an active molecule derived from probiotic Lactobacillus brevis, improves the fibrosis in murine colitis. Transl. Res. 2015, 166, 163–175.
- Park, J.S.; Choi, J. A probiotic complex, rosavin, zinc, and prebiotics ameliorate intestinal inflammation in an acute colitis mouse model. J. Transl. Med. 2018, 16, 37.
- Liu, M.; Zhang, X. Protective effects of a novel probiotic strain, Lactococcus lactis ML2018, in colitis: In vivo and in vitro evidence. Food Funct. 2019, 10, 1132–1145.
- Deng, X.; Zheng, C. Treatment with a probiotic combination reduces abdominal adhesion in rats by decreasing intestinal inflammation and restoring microbial composition. Oncol. Rep. 2020, 43, 986–998.
- Lombardi, F.; Augello, F.R. Soluble Fraction from Lysate of a High Concentration Multi-Strain Probiotic Formulation Inhibits TGF-β1-Induced Intestinal Fibrosis on CCD-18Co Cells. Nutrients 2021, 13, 882.
- Schuppan, D.; Pinzani, M. Anti-fibrotic therapy: Lost in translation? J. Hepatol. 2012, 56, S66–S74.
- Chen, Y.; Lin, Y. Robust bioengineered 3D functional human intestinal epithelium. Sci. Rep. 2015, 5, 13708.
- Giuffrida, P.; Curti, M. Decellularized Human Gut as a Natural 3D Platform for Research in Intestinal Fibrosis. Inflamm. Bowel Dis. 2019, 25, 1740–1750.
- Giuffrida, P.; Pinzani, M. Biomarkers of intestinal fibrosis—One step towards clinical trials for stricturing inflammatory bowel disease. United Eur. Gastroenterol J. 2016, 4, 523–530.
- Steiner, C.A.; Berinstein, J.A. Biomarkers for the Prediction and Diagnosis of Fibrostenosing Crohn’s Disease: A Systematic Review. Clin. Gastroenterol. Hepatol. 2021, 21, S1542–S3565.
- Danese, S.; Bonovas, S. Identification of Endpoints for Development of Antifibrosis Drugs for Treatment of Crohn’s Disease. Gastroenterology 2018, 155, 76–87.