Free fatty acids (FFAs) are metabolically produced and utilized as energy substrates during almost every biological process in the human body. Contrary to long- and medium-chain FFAs, which are mainly synthesized from dietary triglycerides, short-chain FFAs (SCFAs) derive from the gut microbiota-mediated fermentation of indigestible dietary fiber. Originally thought to serve only as energy sources, FFAs are now known to act as ligands for a specific group of cell surface receptors called FFA receptors (FFARs), thereby inducing intracellular signaling to exert a variety of cellular and tissue effects. All FFARs are G protein-coupled receptors (GPCRs) that play integral roles in the regulation of metabolism, immunity, inflammation, hormone/neurotransmitter secretion, etc. Four different FFAR types are known to date, with FFAR1 (formerly known as GPR40) and FFAR4 (formerly known as GPR120) mediating long- and medium-chain FFA actions, while FFAR3 (formerly GPR41) and FFAR2 (formerly GPR43) are essentially the SCFA receptors (SCFARs), responding to all SCFAs, including acetic acid, propionic acid, and butyric acid. As with various other organ systems/tissues, the important roles the SCFARs (FFAR2 and FFAR3) play in physiology and in various disorders of the cardiovascular system have been revealed over the last fifteen years.
- cardiovascular
- FFAR2
- FFAR3
- GPCR
- Signal transduction
1. FFAR2 Signaling and Cardiovascular Function
1.1. Signaling of FFAR2
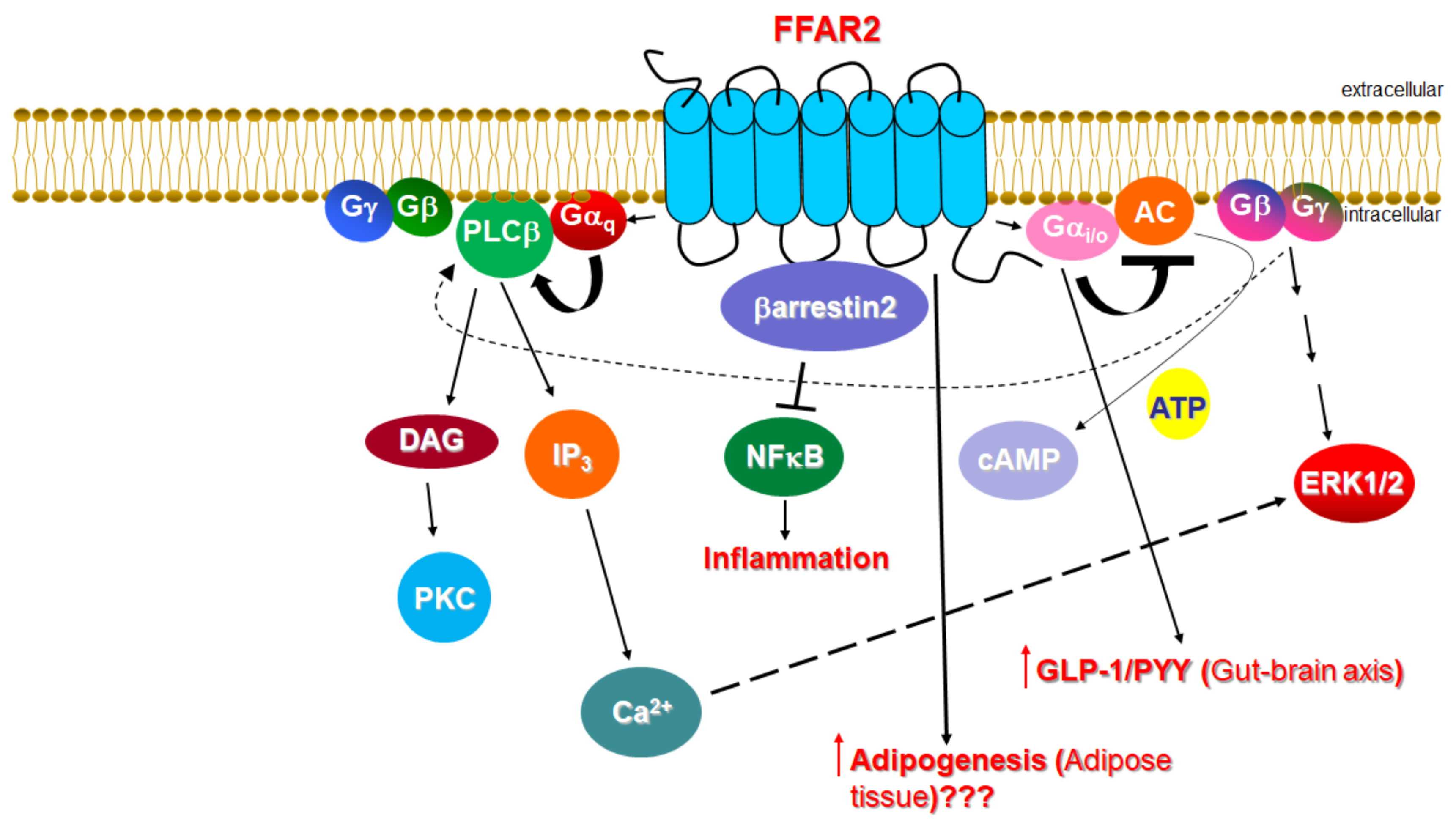
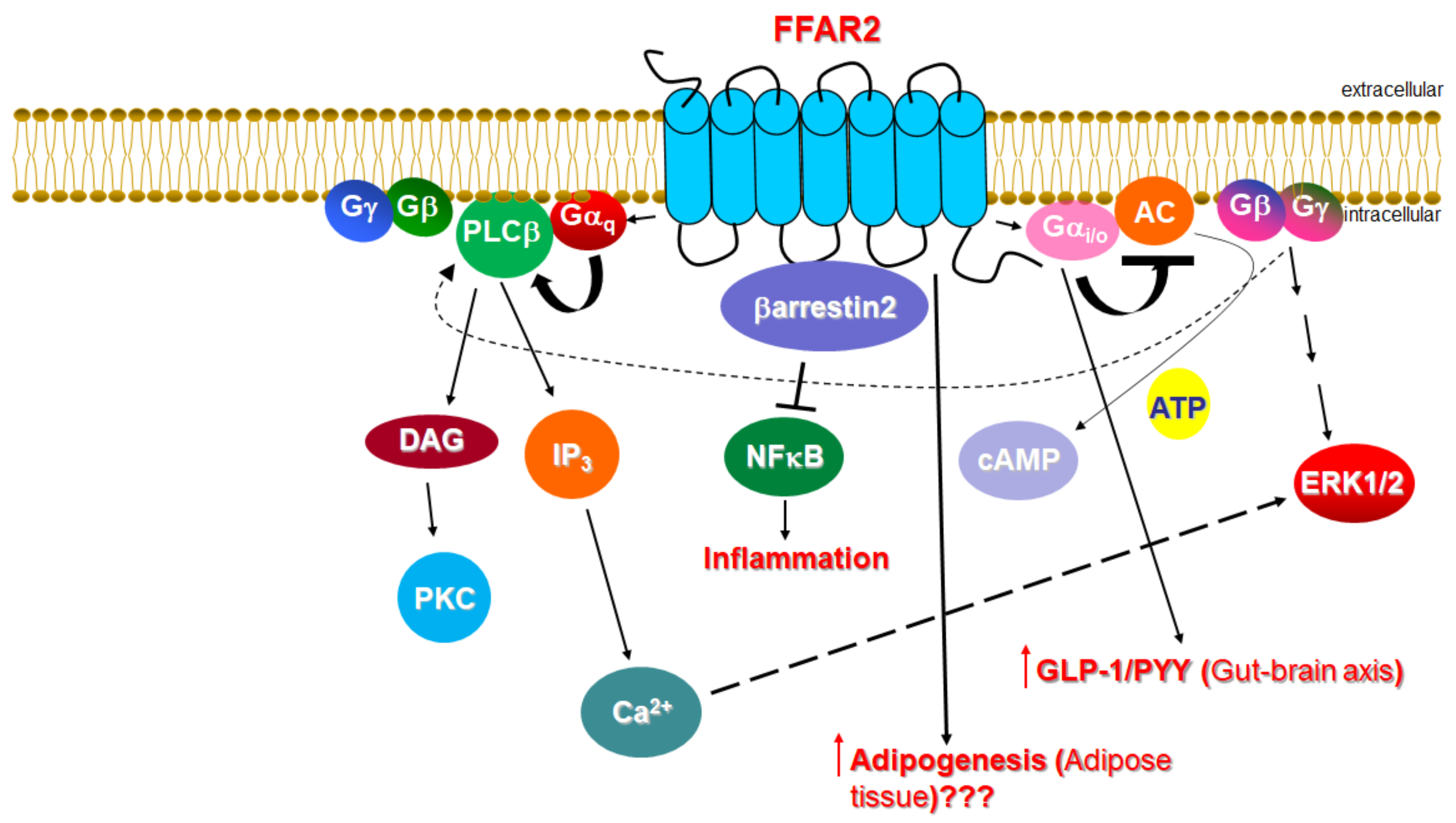
1.2. Function of FFAR2 in Relation to the Cardiovascular System
2. FFAR3 Signaling and Cardiovascular Function
2.1. Signaling of FFAR3
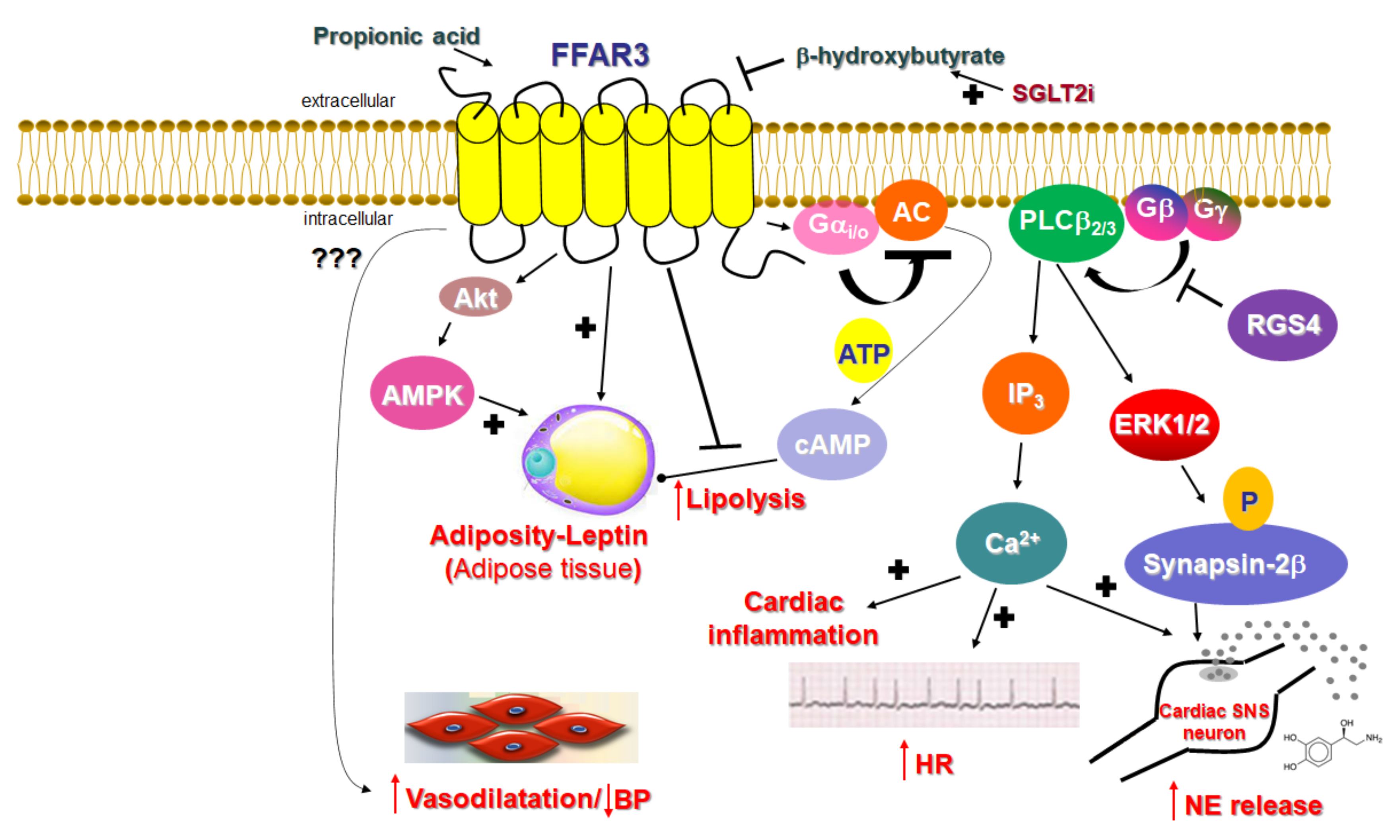
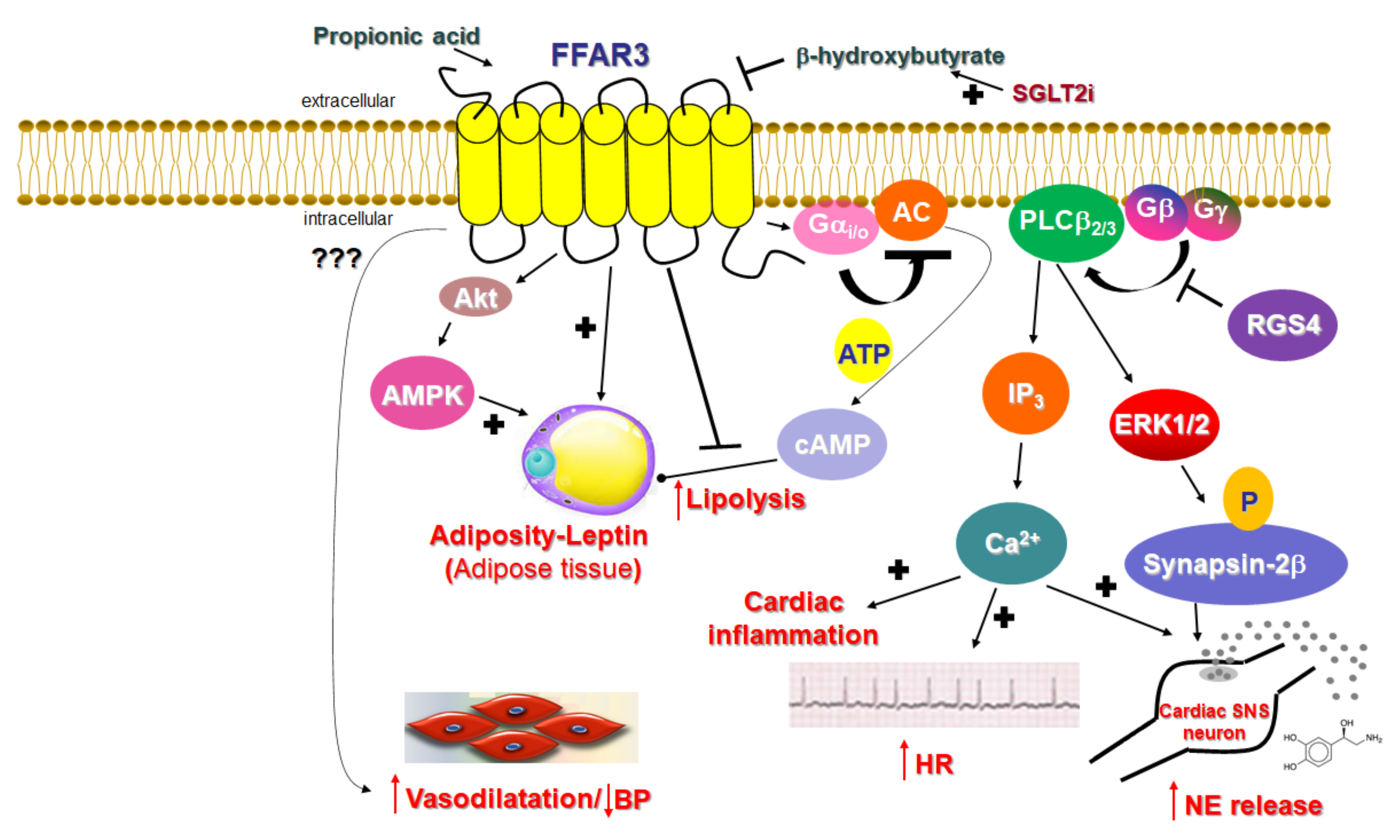
2.2. Function of FFAR3 in Relation to the Cardiovascular System
References
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319.
- Nilsson, N.E.; Kotarsky, K.; Owman, C.; Olde, B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem. Biophys. Res. Commun. 2003, 303, 1047–1052.
- Hudson, B.D.; Tikhonova, I.G.; Pandey, S.K.; Ulven, T.; Milligan, G. Extracellular ionic locks determine variation in constitutive activity and ligand potency between species orthologs of the free fatty acid receptors FFA2 and FFA3. J. Biol. Chem. 2012, 287, 41195–41209.
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210.
- Mishra, S.P.; Karunakar, P.; Taraphder, S.; Yadav, H. Free Fatty Acid Receptors 2 and 3 as Microbial Metabolite Sensors to Shape Host Health: Pharmacophysiological View. Biomedicines 2020, 8, 154.
- Lee, S.U.; In, H.J.; Kwon, M.S.; Park, B.O.; Jo, M.; Kim, M.O.; Cho, S.; Lee, S.; Lee, H.J.; Kwak, Y.S.; et al. β-Arrestin 2 mediates G protein-coupled receptor 43 signals to nuclear factor-κB. Biol. Pharm. Bull. 2013, 36, 1754–1759.
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158.
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G53–G65.
- Thorburn, A.N.; Macia, L.; Mackay, C.R. Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 2014, 40, 833–842.
- Ferrari, F.; Scheffel, R.S.; Martins, V.M.; Santos, R.D.; Stein, R. Glucagon-Like Peptide-1 Receptor Agonists in Type 2 Diabetes Mellitus and Cardiovascular Disease: The Past, Present, and Future. Am. J. Cardiovasc. Drugs 2021.
- Li, S.; Kararigas, G. Role of Biological Sex in the Cardiovascular-Gut Microbiome Axis. Front. Cardiovasc. Med. 2022, 8, 759735.
- Zou, J.; Chassaing, B.; Singh, V.; Pellizzon, M.; Ricci, M.; Fythe, M.D.; Kumar, M.V.; Gewirtz, A.T. Fiber-Mediated Nourishment of Gut Microbiota Protects against Diet-Induced Obesity by Restoring IL-22-Mediated Colonic Health. Cell Host Microbe 2018, 23, 41–53.e4.
- Hong, Y.H.; Nishimura, Y.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Gotoh, C.; Choi, K.C.; Feng, D.D.; Chen, C.; Lee, H.G.; et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 2005, 146, 5092–5099.
- Hu, J.; Kyrou, I.; Tan, B.K.; Dimitriadis, G.K.; Ramanjaneya, M.; Tripathi, G.; Patel, V.; James, S.; Kawan, M.; Chen, J.; et al. Short-Chain Fatty Acid Acetate Stimulates Adipogenesis and Mitochondrial Biogenesis via GPR43 in Brown Adipocytes. Endocrinology 2016, 157, 1881–1894.
- Bjursell, M.; Admyre, T.; Göransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly, Y.M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220.
- Dewulf, E.M.; Ge, Q.; Bindels, L.B.; Sohet, F.M.; Cani, P.D.; Brichard, S.M.; Delzenne, N.M. Evaluation of the relationship between GPR43 and adiposity in human. Nutr. Metab. 2013, 10, 11.
- Frost, G.; Cai, Z.; Raven, M.; Otway, D.T.; Mushtaq, R.; Johnston, J.D. Effect of short chain fatty acids on the expression of free fatty acid receptor 2 (Ffar2), Ffar3 and early-stage adipogenesis. Nutr. Diabetes 2014, 4, 128.
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.Y.; Lannoy, V.; Decobecq, M.E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489.
- Vögler, O.; Barceló, J.M.; Ribas, C.; Escribá, P.V. Membrane interactions of G proteins and other related proteins. Biochim. Biophys. Acta 2008, 1778, 1640–1652.
- Bolognini, D.; Tobin, A.B.; Milligan, G.; Moss, C.E. The Pharmacology and Function of Receptors for Short-Chain Fatty Acids. Mol. Pharmacol. 2016, 89, 388–398.
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035.
- Lymperopoulos, A.; Cora, N.; Maning, J.; Brill, A.R.; Sizova, A. Signaling and function of cardiac autonomic nervous system receptors: Insights from the GPCR signalling universe. FEBS J. 2021, 288, 2645–2659.
- Lymperopoulos, A.; Rengo, G.; Gao, E.; Ebert, S.N.; Dorn, G.W., 2nd; Koch, W.J. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J. Biol. Chem. 2010, 285, 16378–16386.
- Xu, X.; Kaindl, J.; Clark, M.J.; Hübner, H.; Hirata, K.; Sunahara, R.K.; Gmeiner, P.; Kobilka, B.K.; Liu, X. Binding pathway determines norepinephrine selectivity for the human β1AR over β2AR. Cell Res. 2021, 31, 569–579.
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenal adrenoceptors in heart failure: Fine-tuning cardiac stimulation. Trends Mol. Med. 2007, 13, 503–511.
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bucked, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96.
- Inoue, D.; Kimura, I.; Wakabayashi, M.; Tsumoto, H.; Ozawa, K.; Hara, T.; Takei, Y.; Hirasawa, A.; Ishihama, Y.; Tsujimoto, G. Short-chain fatty acid receptor GPR41-mediated activation of sympathetic neurons involves synapsin 2b phosphorylation. FEBS Lett. 2012, 586, 1547–1554.
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev. 2001, 53, 1–24.
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125.
- Squires, K.E.; Montañez-Miranda, C.; Pandya, R.R.; Torres, M.P.; Hepler, J.R. Genetic Analysis of Rare Human Variants of Regulators of G Protein Signaling Proteins and Their Role in Human Physiology and Disease. Pharmacol. Rev. 2018, 70, 446–474.
- Dowal, L.; Elliott, J.; Popov, S.; Wilkie, T.M.; Scarlata, S. Determination of the contact energies between a regulator of G protein signaling and G protein subunits and phospholipase C beta 1. Biochemistry 2001, 40, 414–421.
- Zeng, W.; Xu, X.; Popov, S.; Mukhopadhyay, S.; Chidiac, P.; Swistok, J.; Danho, W.; Yagaloff, K.A.; Fisher, S.L.; Ross, E.M.; et al. The N-terminal domain of RGS4 confers receptor-selective inhibition of G protein signaling. J. Biol. Chem. 1998, 273, 34687–34690.
- Bansal, G.; Druey, K.M.; Xie, Z. R4 RGS proteins: Regulation of G-protein signaling and beyond. Pharmacol. Ther. 2007, 116, 473–495.
- Carbone, A.M.; Cora, N.; Suster, M.S.; Ferraino, K.E.; Borges, J.I.; Nagliya, D.; Sizova, A.; Wang, G.; Iacobellis, G.I.; Goldberger, J.J.; et al. RGS4 Mediates Catecholaminergic Inhibition of Short-Chain Fatty Acid Receptor FFAR3 Signaling & Function in Cardiomyocytes. FASEB J. 2022, 36, 2.
- Colina, C.; Puhl, H.L., 3rd; Ikeda, S.R. Selective tracking of FFAR3-expressing neurons supports receptor coupling to N-type calcium channels in mouse sympathetic neurons. Sci. Rep. 2018, 8, 17379.
- Kaji, I.; Akiba, Y.; Konno, K.; Watanabe, M.; Kimura, S.; Iwanaga, T.; Kuri, A.; Iwamoto, K.; Kuwahara, A.; Kaunitz, J.D. Neural FFA3 activation inversely regulates anion secretion evoked by nicotinic ACh receptor activation in rat proximal colon. J. Physiol. 2016, 594, 3339–3352.
- Tough, I.R.; Forbes, S.; Cox, H.M. Signaling of free fatty acid receptors 2 and 3 differs in colonic mucosa following selective agonism or coagonism by luminal propionate. Neurogastroenterol. Motil. 2018, 30, e13454.
- Nakagawa, Y.; Kuwahara, K. Sodium-Glucose Cotransporter-2 inhibitors are potential therapeutic agents for treatment of non-diabetic heart failure patients. J. Cardiol. 2020, 76, 123–131.
- Lymperopoulos, A.; Borges, J.I.; Cora, N.; Sizova, A. Sympatholytic Mechanisms for the Beneficial Cardiovascular Effects of SGLT2 Inhibitors: A Research Hypothesis for Dapagliflozin’s Effects in the Adrenal Gland. Int. J. Mol. Sci. 2021, 22, 7684.
- Uthman, L.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Kuschma, M.C.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Direct Cardiac Actions of Sodium Glucose Cotransporter 2 Inhibitors Target Pathogenic Mechanisms Underlying Heart Failure in Diabetic Patients. Front. Physiol. 2018, 9, 1575.
- Nøhr, M.K.; Egerod, K.L.; Christiansen, S.H.; Gille, A.; Offermanns, S.; Schwartz, T.W.; Møller, M. Expression of the short chain fatty acid receptor GPR41/FFAR3 in autonomic and somatic sensory ganglia. Neuroscience 2015, 290, 126–137.
- Nøhr, M.K.; Pedersen, M.H.; Gille, A.; Egerod, K.L.; Engelstoft, M.S.; Husted, A.S.; Sichlau, R.M.; Grunddal, K.V.; Poulsen, S.S.; Han, S.; et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 2013, 154, 3552–3564.
- Shimizu, H.; Masujima, Y.; Ushiroda, C.; Mizushima, R.; Taira, S.; Ohue-Kitano, R.; Kimura, I. Dietary short-chain fatty acid intake improves the hepatic metabolic condition via FFAR3. Sci. Rep. 2019, 9, 16574.
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371.
- Jocken, J.W.E.; González Hernández, M.A.; Hoebers, N.T.H.; van der Beek, C.M.; Essers, Y.P.G.; Blaak, E.E.; Canfora, E.E. Short-Chain Fatty Acids Differentially Affect Intracellular Lipolysis in a Human White Adipocyte Model. Front. Endocrinol. 2018, 8, 372.
- Ohira, H.; Fujioka, Y.; Katagiri, C.; Mamoto, R.; Aoyama-Ishikawa, M.; Amako, K.; Izumi, Y.; Nishiumi, S.; Yoshida, M.; Usami, M.; et al. Butyrate attenuates inflammation and lipolysis generated by the interaction of adipocytes and macrophages. J. Atheroscler. Thromb. 2013, 20, 425–442.
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772.
- Bellahcene, M.; O’Dowd, J.F.; Wargent, E.T.; Zaibi, M.S.; Hislop, D.C.; Ngala, R.A.; Smith, D.M.; Cawthorne, M.A.; Stocker, C.J.; Arch, J.R. Male mice that lack the G-protein-coupled receptor GPR41 have low energy expenditure and increased body fat content. Br. J. Nutr. 2013, 109, 1755–1764.
- Zaibi, M.S.; Stocker, C.J.; O’Dowd, J.; Davies, A.; Bellahcene, M.; Cawthorne, M.A.; Brown, A.J.; Smith, D.M.; Arch, J.R. Roles of GPR41 and GPR43 in leptin secretory responses of murine adipocytes to short chain fatty acids. FEBS Lett. 2010, 584, 2381–2386.