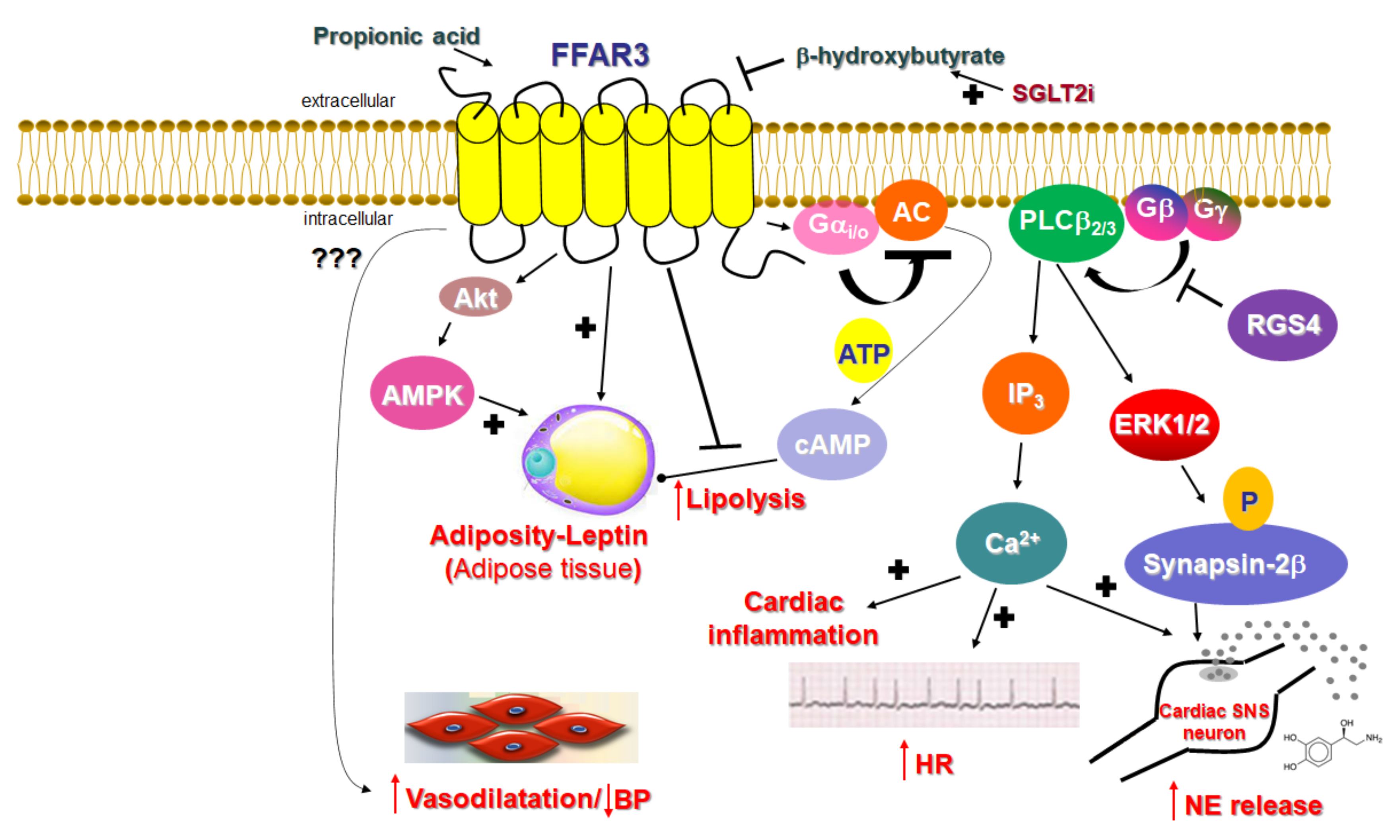
Unlike FFAR2, whose role in cardiovascular homeostasis (if any), as mentioned above, is virtually unknown, FFAR3’s involvement in cardiovascular function regulation has become increasingly clear over the past decade. The main physiological mechanism by which FFAR3 regulates cardiac function is via the effects it exerts in the sympathetic nervous system
[21]. FFAR3 is robustly expressed in murine peripheral sympathetic neurons, including cardiac sympathetic nerve terminals, wherein it regulates whole body metabolic homeostasis, along with neuronal activity/firing by inducing norepinephrine release
[21] (
Figure 2). Although both norepinephrine and epinephrine mediate the effects of the sympathetic nervous system on all cells and tissues of the entire body, norepinephrine is the actual neurotransmitter synthesized, stored, and released from sympathetic neurons
[22][23][24][77,78,79]. Epinephrine is the hormone synthesized in the adrenal medulla and secreted into the systemic circulation
[22][23][24][77,78,79]. This is because sympathetic neurons lack the enzyme phenyl-ethanolamine-N-methyltransferase (PNMT), which converts norepinephrine to epinephrine
[23][25][78,80]. FFAR3 is also present in portal neurons of the liver, where it regulates propionate-induced gluconeogenesis via the gut-brain axis
[26][81]. FFAR3 knockout mice display significantly lower catecholamine (norepinephrine and epinephrine) synthesis, as evidenced by the downregulation of tyrosine hydroxylase, the enzyme that catalyzes the rate-limiting step of catecholamine biosynthesis
[21] (
Figure 2). Consistent with a lower sympathetic neuronal activity/firing rate, heart rate is also reduced in FFAR3 knockout mice
[21] (
Figure 2). Thus, FFAR3 clearly promotes neuronal firing and norepinephrine synthesis and release in sympathetic neurons. The signaling pathway underlying this effect of FFAR3 is the stimulation of the Gi/o-derived free Gbg subunit activation of PLCβ
2/3 (see above)
[21]. G
βγ-activated PLCβ
2/3 activates, in turn, the MAPKs ERK1/2, which phosphorylate synapsin-2β at Ser426 to induce vesicle fusion with the neuronal plasma membrane and norepinephrine exocytosis/synaptic release from sympathetic nerve endings
[27][82] (
Figure 2). Notably, neither GRK2 nor β-arrestins appear to be involved in this signaling pathway
[21], although GRK2 should theoretically play a role, since it interacts with free Gβγ subunits via its C-terminal pleckstrin homology (PH) domain
[28][54]. In fact, this is the main mechanism for membrane targeting and the activation of GRK2 (and GRK3)
[29][55]. On the other hand, RGS proteins of the B/R4 family, which inactivate Gi/o proteins, must interfere with this FFAR3-dependent signaling pathway in sympathetic neurons
[30][53]. RGS4 in particular is known to directly bind Gi/o-derived free G
βγ subunits and PLCβ and to inhibit PLCβ activation independently of its RGS function
[31][32][33][83,84,85]. Indeed, while studying FFAR3 signaling and function in rat cardiomyocytes, researchers have confirmed that RGS4 intervenes in FFAR3 signaling to PLCβ via the Gi/o-derived free G
βγ subunits, dampening the subsequent PLCβ-induced Ca
2+ signaling from this receptor and leading to inflammation in the heart, as well as norepinephrine release and firing activity in cardiac sympathetic neurons
[34][86] (
Figure 2). Nevertheless, other studies have suggested that FFAR3 may actually inhibit secretion/exocytosis via N-type Ca
2+ channel inhibition in enteric and vascular neurons
[35][36][87,88]. More specifically, FFAR3 signaling inhibits N-type Ca
2+ channels via G
βγ signaling, reducing neuronal catecholamine release in rat sympathetic neurons innervating vascular smooth muscle
[35][87]. Additionally, FFAR3 modulates the cholinergic-mediated secretory response in the proximal colonic mucosal neurons of rats
[36][88], and FFAR3 is a putative target for neurogenic bowel disorder treatment
[37][89]. Indeed, the FFAR3 synthetic agonist AR420626 suppresses cholinergic and serotonergic-dependent colonic motility and secretions
[37][89]. Therefore, the picture regarding neuronal FFAR3 effects is undoubtedly complicated, and more studies are required to provide better clarity. The decade-old study by Kimura et al. in FFAR3 knockout mice demonstrated that the FFAR3-dependent norepinephrine release from sympathetic neurons modulates energy expenditure and that the activation of FFAR3 with propionic acid elevates heart rate and increases cardiac oxygen demand/consumption
[21]. It also showed that the effect of propionate/FFAR3 on heart rate was suppressed by pretreatment with a β-adrenergic receptor (AR) blocker, which indicated that FFAR3 signaling is reciprocally regulated by βARs, i.e., there is a signaling crosstalk between FFAR3 and βARs, upregulating βAR function in sympathetic ganglions
[21]. Notably, in that same study, the ketone body β-hydroxybutyrate (or 3-hydroxybutyrate) was shown to block FFAR3 and antagonize its pro-sympathetic hyperactivity in neurons
[21] (
Figure 2). On the other hand, sodium/glucose co-transporter (SGLT)-2 inhibitors, such as dapagliflozin and empagliflozin (anti-diabetic/diuretic drugs with a plethora of beneficial cardiovascular effects that have been coming to light at an accelerating pace), increase ketone body (including β-hydroxybutyrate) production in the heart and blood vessels
[38][39][90,91]. Given that dapagliflozin and empagliflozin have been shown to possess sympatholytic properties that mediate, at least in part, their beneficial effects in chronic human heart failure
[40][92], it is tempting to speculate that the sympatholytic effects of SGLT2 inhibitor drugs are mediated, at least partially, by the b-hydroxybutyrate-mediated blockade of FFAR3 signaling in sympathetic neurons, which normally raises cardiac norepinephrine levels and cardiovascular sympathetic nervous system activity
[39][91]. This, of course, awaits confirmation by future studies in experimental models of chronic heart failure.
FFAR3 is expressed, not only in postganglionic sympathetic and sensory neurons of the autonomic nervous system, but also in sympathetic and sensory neurons of the somatic peripheral nervous system
[21][35][41][21,87,93]. Thus, SCFAs exert their effects via FFAR3 not only through the enteroendocrine system, but also directly by modifying physiological reflexes integrating the peripheral nervous system and the gastrointestinal tract. Moreover, FFAR3 in submucosal, and the myenteric ganglionic plexus neurons of the small intestine regulate gut hormonal synthesis, including GLP1 and PYY synthesis, similar to FFAR2 (see above)
[42][43][44][3,5,11]. Additionally, FFAR3 significantly reduces lipolysis by inhibiting hormone-sensitive lipase phosphorylation and activity via Gαi
-mediated AC inhibition and cAMP lowering in peripheral adipose tissues
[45][46][15,94] and increases leptin production, hepatic lipogenesis, and adipocyte growth
[47][4] (
Figure 2). Indeed, male FFAR3 knockout mice treated with HFD exhibit more body fat mass and higher blood glucose levels compared to wild type female littermates
[48][95], and leptin synthesis is reduced in FFAR3 knockouts
[49][96].