Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Amaresh K Ranjan and Version 3 by Beatrix Zheng.
Neurological/neurovascular disorders constitute the leading cause of disability and the second leading cause of death globally. Major neurological/neurovascular disorders or diseases include cerebral stroke, Alzheimer’s disease, spinal cord injury, neonatal hypoxic-ischemic encephalopathy, and others. Their pathophysiology is considered highly complex and is the main obstacle in developing any drugs for these diseases. Studies have demonstrated the highly promising effects of sovateltide on the neurovascular system and improved recovery, comprehensively after various injuries/insults, which have generated immense hope of developing an effective therapy with the potential to treat various neurological disorders.
- endothelin B receptors
- neurological diseases
- neurovascular disorders
- sovateltide
- IRL-1620
- neurogenesis
- stem/progenitor cells
- regeneration
1. Stroke
Cerebral stroke due to perturbation or blockage of blood flow in brain tissues leads to damage or death of neuronal cells and causes morbidity and mortality in patients. According to recent statistical data, stroke is the second leading cause of death worldwide, with an annual mortality of 5.5 million [1]. Data also suggest that ischemic stroke is more predominant, with ~80% of stroke cases being of the ischemic type [2]. Despite high mortality, treatment of stroke remains challenging. At present, only one FDA-approved drug for ischemic stroke, the thrombolytic agent, rtPA or Alteplase, is available. However, its use is limited due to a short therapeutic window of 3–4.5 h from the onset of symptoms and also has a risk of intracranial hemorrhage in 2–7% of patients [3]. There is an urgent need to develop new effective drugs for stroke to alleviate neurological deficit and repair cerebral damage. However, due to the complex pathophysiology of stroke, the development of new drugs seems quite challenging, as evident by the failure of several drugs in their late stages of clinical trials [4]. Cerebral ischemic pathophysiology involves hypoxia, vascular damage, inflammation, apoptosis, and other events, which cause neuronal cell damage/death and functional impairment in the brain. Significant efforts are being made to understand the complex pathophysiology of stroke, and various mechanisms, e.g., anticoagulation, neuroprotection, and neuroregeneration, are being explored [5]. Interestingly, endothelin (ET) signaling in the mammalian neurovascular system regulates these important pathophysiological events. Therefore, ETs and their receptors, endothelin A (ETARs) and endothelin B (ETBRs)ETARs and ETBRs, are considered one of the most potent targets for developing new drugs for stroke.
Studies have shown an elevated level of ET-1 in blood and brain tissues following cerebral ischemia and correlated with the tissue damage in the ischemic brain [6][7]. Since ET-1 primarily binds to ETARs and acts as a vasoconstrictor, vasoconstriction and associated events were presumed to be the leading cause of cerebral tissue damage, and it was hypothesized that ETARs antagonists would reduce the damage. Therefore, several studies focused on testing the effect of various ETARs inhibitors such as BQ123, SB234551, A-127722, and S-1039 were performed and demonstrated a reduction in neural tissue damage and inflammation and neurological deficits following cerebral ischemia [8][9][10][11][12]. However, unfortunately, none of these could advance to the clinical testing level. Similarly, studies performed using combined ETARs/ETBRs receptors antagonists, TAK-044, bosentan and SB209670 showed mixed results as follows: TAK-044 was effective in decreasing oxidative stress and ischemia, whereas bosentan and SB209670 had no significant effect on improving the condition in animal models of acute ischemic stroke [13][14].
Interestingly, on the other hand, antagonizing ETBRs had deleterious effects and caused poor outcomes [15][16]. These observations suggested the critical roles of ETBRs in protecting brain tissues from ischemic damage following a cerebral stroke. Hence, testing the effect of enhanced ETBRs signaling in ischemic cerebral tissues using specific agonists would be useful for discovering novel potent drugs to treat ischemic stroke.
Studies have shown that ETBRs play an important role in neural cell survival and proliferation [17][18][19]. Normally, expression of ETBRs in the adult is observed on vascular endothelial and smooth muscle cells; however, under pathological conditions, it is also observed in NPCs (neural progenitor cells), NPs (neuronal progenitors), GPs (glial progenitors), and glial cells of the central nervous system (CNS) [20][21][22]. Moreover, they also play an essential role in the development of CNS. The rodent model deficient in ETBRs during the prenatal period had CNS disturbances and fatal congenital anomalies. The deficiency of ETBRs in the postnatal stage is known to cause increased apoptosis and decreased NPs population in different regions of the adult brain [17][23][24]. Besides their importance in brain development, thwe researchers hhave demonstrated the role of ETBRs in the repair and regeneration of adult brains after stroke [5][25][26]. ThWe researchers sstimulated ETBRs by intravenous administration of a selective agonist sovateltide in permanent middle cerebral artery occluded (MCAO) rats. The researchers We observed significantly improved neurological and motor functions and several indications of repair and regeneration, including a decrease in infarct volume and oxidative stress, increased pro-angiogenic, pro-survival, and anti-apoptotic markers, and an increased number of proliferating cells in the brain [27][28][29][30][31][32]. However, these improvements were abrogated by BQ788, an antagonist of ETBRs, which proved the role of sovateltide mediated selective stimulation of ETBRs in these improvements. These results showed the role of sovateltide in providing protection and functional improvement of neural cells in the stroked brain. Some of the remarkable preclinical studies in MCAO rats have demonstrated the roles of sovateltide in enhancing cerebral blood flow and decreasing apoptosis of neural cells after ischemic stroke in the brain [28][29][31][32]. In theiour recent research, theystudy, we observed increased phospho-Akt level and decreased Bad expression at 7 h post-MCAO, higher level of anti-apoptotic Bcl-2, and lower level of pro-apoptotic Bax in sovateltide treated MCAO rats than control MCAO rats. The researchstudy also showed significantly decreased mitochondrial membrane-bound Bax intensity in sovateltide compared to control MCAO rats at days 1 and 7 post-MCAO [31], which indicated inhibition of mitochondrial apoptotic pathway by sovateltide. The cell damage was assessed by TUNEL assay in these rats, which showed lower cell damage in sovateltide treated rats on day 1 and day 7. Moreover, significantly improved neurological and motor functions in these rats were observed after sovateltide treatment [31]. Since the improvement in these functions is known to be associated with the fate of mitochondria and their activity, in another research, the researchers etudy, we examined mitochondrial fusion, fission, morphology, and biogenesis in sovateltide treated MCAO rat brains. The researchers We observed improved mitochondrial fusion, decreased fission, increased size and biogenesis in sovateltide treated rat brain tissues [33]. ThWe researchers aalso studied the role of sovateltide mediated ETBRs agonism in the regeneration of neuronal cells in adult MCAO rat brains [33][34] and showed a novel role of sovateltide in neuronal progenitor cells (NPCs) mediated regeneration and repair in MCAO rat brains. Ischemic stroke was induced in the right cerebral hemisphere of adult rats using the MCAO technique, and rats were treated with sovateltide (5 µg/kg body wt.) at 4, 6, and 8 h on day 0, 3, and 6 post-MCAO. They were sacrificed at 24 h or at day 7 post-MCAO, and tissues from the right (RH) and left (LH) cerebral hemispheres were analyzed for expression of neural progenitor markers (DCX, HuC/HuD, and NeuroD1) and stem cell markers (Oct-4 and Sox-2). Significant upregulation of DCX and HuC/HuD in RH, while NeuroD1 in both RH and LH was observed 24 h post-MCAO in sovateltide treated rats. On the other hand, insignificant change in the expression of multipotent stem cell markers, Oct4 and Sox-2 was observed in these rats. Sovateltide treated rats also significantly decreased in the infarct area at day 7 post-MCAO [34].
To explore the role of sovateltide on NPCs differentiation, thwe researchers ccultured adult rat NPCs and exposed them to in vitro hypoxia. NPCs exposed to sovateltide showed significantly higher expression of neuroD1 and NeuN than cells exposed to the vehicle [33]. These results indicated that sovateltide promoted differentiation/maturation of NPs to generate a mature neuronal cell population, which would heal the stroked brain more efficiently (Figure 1). These findings have demonstrated a novel mechanism of action of sovateltide, which promotes differentiation of NPs in the ischemic stroked brain and helps in neural regeneration and repair. No other agents in the pipeline of drug development for stroke have shown such type of mechanism of action, and therefore sovateltide has the potential to be developed as a novel first-in-class drug for ischemic stroke.
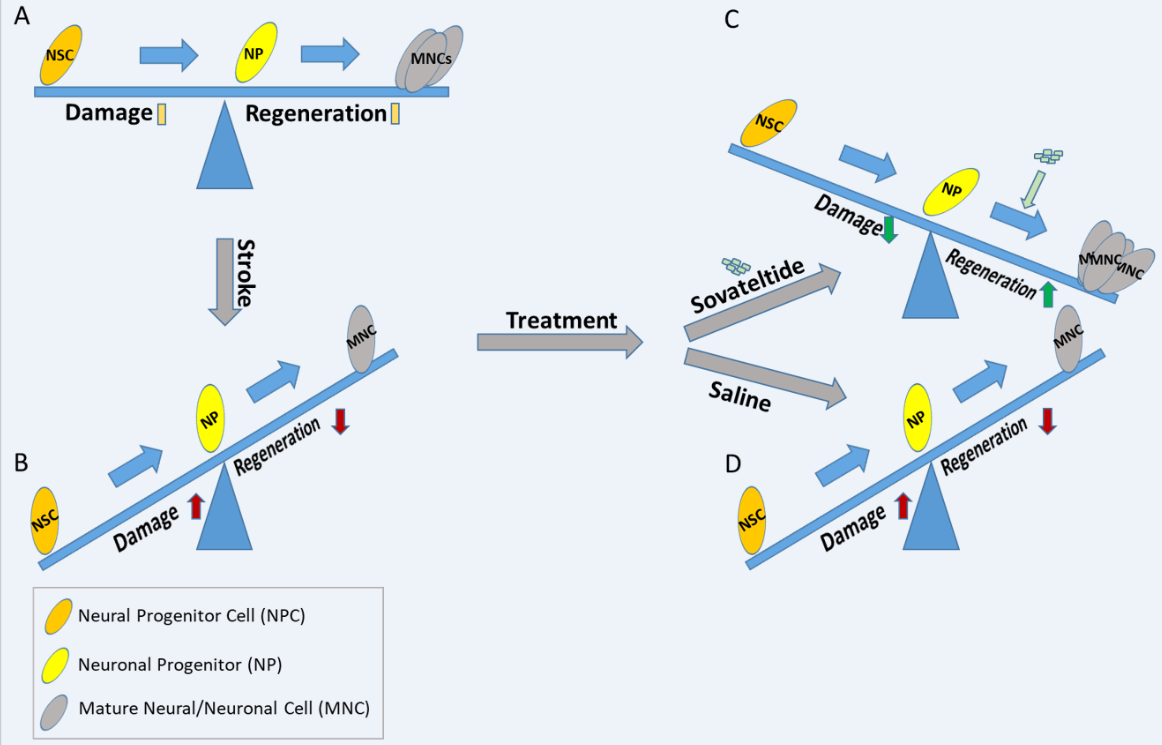
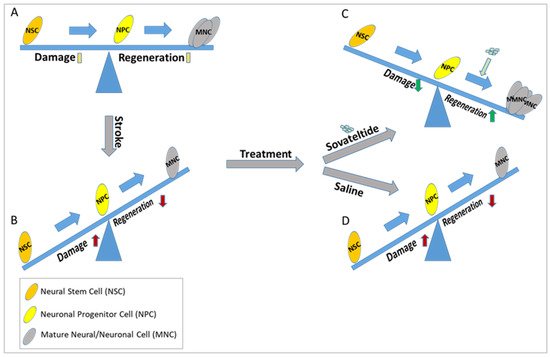
Figure 1. Diagrammatic representation of the effect of sovateltide on neuronal progenitor cell differentiation and neural regeneration after cerebral ischemic stroke. (A) Normal homeostasis, (B) Higher damage than regeneration (probably due to less differentiation of NPCs), (C) Higher regeneration and lower damage after sovateltide treatment (probably due to higher NPCs differentiation), which remains similar after saline treatment (D).
2. Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive loss of neuronal cells and cognitive decline and is also associated with aging [35]. According to a recent report, approximately 5.8 million Americans of all ages were suffering from AD in 2019, which is projected to rise to 14 million by 2050 [36]. The cost in 2019 for all individuals with AD and other dementias was approximately USD 277 billion. Compared to other diseases such as stroke and heart failure, the proportion of deaths related to AD is going up, and it increased by 89% between 2000 and 2014 [37][38]. At present, the management and care of AD rely only on two classes of pharmacologically approved therapies: first, cholinesterase inhibitors, and second, N-methyl-D-aspartate receptor antagonists. The cholinesterase inhibitors donepezil, rivastigmine, and galantamine are recommended therapy for patients with mild, moderate, or severe AD dementia [38]. On the other hand, memantine is approved for use in moderate-to-severe AD patients. It has antagonistic activity on N-methyl-D-aspartate receptor and agonistic activity on dopamine. Although the existing drugs help in concealing AD symptoms, they fail to cure or delay AD progression. Some of the pathological hallmarks of AD include β-amyloid plaque deposition and neurofibrillary tangles of hyperphosphorylated tau protein [39][40]. The neuropathology involved in AD is highly complex, making drug development very difficult, as evident by failing most of the AD drug trials in the past. Approximately, 413 AD trials were performed from 2002 to 2012 but 99.6% failed and could not meet the endpoints [41]. One of the reasons for the failure of these trials could be their reliability on the most common hallmarks of the AD pathology, beta-amyloid (Aβ) and tau, which are still debatable whether they have any causative role in AD pathology. At present, immunotherapy targeting the clearance of Aβ fibrils and plaques is being developed, which will help in understanding the role of AD hallmarks in the disease. Some of the drugs in this category are aducanumab, donanemab, bapineuzumab, solanezumab, gantenerumab, crenezumab, and ponezumab (PMID: 24959143, PMID: 33720637). Unfortunately, the clinical trials of aducanumab (Aduhelm) failed to demonstrate its efficacy in slowing memory loss or cognitive decline. However, its controversial approval using the FDA’s Accelerated Approval Pathway, based on Aβ plaque clearance, which was a surrogate endpoint of the trial has deeply engraved the AD drug development [42][43][44]. Hence, for developing effective AD drugs, more research in the future focused on examining the neural pathophysiological pathways essential for neuronal cell survival, regeneration, and functional improvement in adult brains are required. Several studies carried out by the ouresearch group and others have shown that the ET system is involved in pathways related to neural cell survival, regeneration, and repair of adult brains after damage [28][29][30][31][33][34][45]. Therefore, testing its potential as a drug target in AD would be useful in developing a new drug, which could repair the damaged area in the brain and restore the cognitive ability and memory of AD patients.
Several studies have demonstrated the involvement of the ET system in AD and provided the following evidence. Significantly elevated levels of ECE-2 and ET-1 have been correlated with AD disease progression and in vitro and in vivo studies have demonstrated their abnormal production because of Aβ [46]. Gene mutation analysis researchtudy has shown increased production of Aβ1–40 and Aβ1–42 after deletion mutations in either ECE-1 or ECE-2 in mouse brains [47][48]. Mice with ECE-2 knockout were observed with impaired learning and memory [49]. Thus, studies have demonstrated a direct link between ETs and AD pathogenesis, and a relationship between ECEs and Aβ protein turnover in the adult brain [46][50]. Apart from the above-mentioned direct links, AD development and progression are also associated with regionally reduced cerebral blood flow and vascular dysfunction [51], which are regulated by ETs [19][52][53]. The ET-1 mediated vasoconstriction in middle cerebral and basilar arteries has been found to be increased following exposure to Aβ [54], also an elevated level of ET-1 has been detected in the post-mortem brains of individuals with AD compared to non-AD individuals. These observations indicated the role of ET-1 in neuronal dysfunction in the early stage of AD development and progression.
ThWe researchers initially hypothesized a potential benefit of ETARs antagonists to reduce the ET-1 mediated effects in the AD brain, which could be developed as novel agents for AD treatment. The researchOur group examined the effect of ETARs antagonists on Aβ-induced neuronal damage and cognitive dysfunction in an adult rat model of AD. The researchers We used BQ123 and BMS182874, specific ETARs antagonists, in theiour studies. Both BQ123 and BMS182874 significantly improved the spatial memory deficit and reduced oxidative damage in the brain caused by Aβ. A significantly reduced escape latency and increased preference for the target quadrant were observed in BQ123, or BMS182874 treated AD rats. However, rats treated with a nonspecific ETARs/ETBRs receptors antagonist, TAK-044, had no improvement in spatial memory deficit [55]. Lack of improvement with nonspecific ETARs/ETBRs antagonist indicated a role of ETBRs signaling in the AD brain. Since ET-1 could bind to both ETARs and ETBRs receptors, normally ETBRs might be countering the action of ETARs signaling; however, in the presence of ETARs antagonists, they would be potentiating their signals. These results suggested the important roles of ETBRs receptors in AD development and progression and could be an appropriate target for AD drug development. ETBRs have shown anti-apoptotic activity in the protected cells from neurotoxicity of Aβ in cultured neurons [23][27][5][56]. Cortical neural progenitor cells showed a high level of ETBRs, and their stimulation leads to proliferation and migration, indicating that stimulation of ETBRs enhances neuroregeneration by directly acting on neural progenitors [17][57][58]. Stimulation of ETBRs is also known to elicit vasodilatation, and previous studies in theiour laboratory have demonstrated that intravenous administration of sovateltide (an ETBRs agonist) increased cerebral blood flow in normal rats [59] and that the expression of an anti-apoptotic marker, Bcl-2, was found to be increased, and pro-apoptotic marker, Bax was found to be decreased in the neuronal cell line, PC-12 [60]. While, in a rat model of AD produced by injecting Aβ1–40 intracerebroventricularly, sovateltide (5 μg/kg body wt.) treatment resulted in a significant functional recovery. ThWe researchers eexamined their behavioral and spatial memory impairments using a Morris Water Maze. AD rats showed impairment in spatial memory as evidenced by significantly longer escape latencies and no preference for the quadrant, which previously contained the platform in the probe trial [61], while AD rats treated with sovateltide significantly reduced the spatial memory deficit caused by Aβ. The Aβ plaque is known to induce oxidative damage to neural cells, and the rwesearchers examined whether sovateltide treatment had any effect on the oxidative stress in the brain of AD rats. Sovateltide treated rats showed significantly decreased levels of malondialdehyde (MDA) and increased levels of reduced glutathione (GSH) and superoxide dismutase (SOD), which indicated significantly reduced oxidative stress in AD rats after sovateltide treatment. However, these improvements were blocked when animals were administered with ETBRs antagonist BQ788, which confirms the effects were due to selective stimulation of ETBRs by sovateltide in these AD rats [61].
The Ouresearchers' other studies related to cerebral stroke in the MCAO rat model of stroke have shown that sovateltide treatment could increase NGF and VEFG expression in rat brains after ischemia [62]. Since these factors are important for neurogenesis, an increase in the expression of these markers suggested the role of ETBRs in neurogenesis in damaged brain tissues. Therefore, thwe researchers treated an APP/PS1 mouse AD model with sovateltide and assessed neurogenesis in the AD mouse brain. The treated mice were observed with a significantly increased expression of neural progenitor markers, NeuroD1 and Doublecortin (DCX), along with elevated expression of nuclear NeuN (a marker for mature neurons) in the mouse brain. These observations suggested an increase in neuronal progenitors and their differentiation to produce mature neuronal cells expressing NeuN in sovateltide treated mice, indicating a higher potential for neuronal regeneration. The synapse formation by newly produced cells in the brain is a prerequisite for its function restoration; hence the researchers aswe assessed the expression of pre-synaptic (synapsin1 and synaptophysin) and post-synaptic marker (PSD95) in these mice. Sovateltide significantly increased the expression of these markers in AD mouse brain tissues. The assessment of learning and memory in control AD mice showed significant impairment in spatial memory as evident by significantly longer escape latencies and no preference for the quadrant which previously contained the platform in the probe trial. On the other hand, AD mice treated with sovateltide had significantly reduced (45, 40, and 46%) learning and memory deficit in 6, 9, and 12 months of age.
Overall, theseour studies have demonstrated that ETBRs agonist, sovateltide is an effective agent, which can significantly reduce the AD-associated neurodegeneration and help in neuronal regeneration and cerebral function recovery by promoting neuro- and synapto-genesis.
3. Spinal Cord Injury
The spinal cord with the brain constitutes the central nervous system. It is a long bundle of nerves and cells which extends from the brain to the lower back. It is required to carry signals to and from the brain, including nerve impulses for movement, sensation, pressure, temperature, pain, and many more. Moreover, it is also known to act independently of the brain in conducting motor reflexes. Thus, the spinal cord plays a vital role in the body’s functioning. The damage to the spinal cord is known as spinal cord injury (SCI), which causes temporary or permanent changes in its function. The SCI etiologies could be traumatic or non-traumatic; however, more than 90% of SCI-reported cases are traumatic, caused by tragic incidences such as traffic accidents, violence, sports, or falls [63]. In traumatic SCI, the primary injury damages tissues in the cord and leads to a highly complex secondary injury cascade, including ischemia, inflammation, and the death of neurons and glial cells. Formation of glial scar and cystic cavities are followed, and consequently, changes in the organizational and structural architecture of the spinal cord are resulted, which may cause permanent neurological deficits. The secondary injury also includes hyperinflammatory and cytotoxic conditions, which further damage neuronal cells and inhibit regeneration and repair processes in the damaged spinal cord.
Moreover, the regenerative capacity in the spinal cord is limited because of an abysmal number of neural progenitor cells and restricted plasticity, which makes the intrinsic recovery potential of the spinal cord very poor and rare [64]. Nonetheless, existing therapeutic approaches include early surgery, strict blood pressure control, and treatment with steroids, which are still debatable for their effect, and they are primarily focused on mitigating secondary injury of SCI, not to cure. Hence, currently available SCI interventions have failed to improve the SCI treatment outcomes. Therefore, finding a cure for SCI is urgently required otherwise, it’s devastating physical, social, and vocational consequences for patients and their families would be continued. In the past four decades, numerous therapies aimed to improve neuroprotection and neurodegeneration have shown little promise from the preclinical to the clinical stage of development. However, further research aimed to understand the complex pathophysiological cascade and neuroregeneration and repair in SCI are required to overcome the limitations in translating the SCI research data obtained from animal models to clinical trials.
The SCI clinical trials carried out so far have explored various approaches, including pharmacologic, cell-based, physiologic, and rehabilitation, to reduce secondary injury and overcome barriers of neuroregeneration and recovery. Undoubtedly, several clinical trials have been completed and are currently being carried out; however, discovering a potent drug to cure SCI still looks beyond reach; a brief detail about these trials is described by Donovan et al. [65]. Nonetheless, a tailored treatment plan combining many of these strategies is being emphasized, which would offer significant benefits for persons with SCI. Since regeneration and repair of tissues after damage in organs is the most important for their functional restoration, thwe researchers believe that the success of the SCI treatment strategy would depend upon how efficiently the researchers cwe could promote the regenerative process in the damaged spinal cord. Hence, the researchers swe suggest spinal regenerative research should be at the core of every combinatorial tailored plan of SCI treatment, and therefore more research in this direction should be promoted to discover novel spinal cord regenerative therapeutics.
TheOur research work focused on regeneration and repair of adult brains after stroke and Alzheimer’s disease by utilizing sovateltide to elicit ETBRs in the brain is highly promising, as described in the above sections of the review. The preclinical and clinical studies of sovateltide in these diseases have created hope for developing sovateltide as a first-in-class therapeutic. It works uniquely by promoting differentiation of neuronal progenitor cells and reducing apoptosis and oxidative stress besides increasing blood flow in the damaged brain tissues and helping in protecting cerebral tissues from damage and promoting regeneration and repair in the damaged tissues. The spinal cord is also a part of the CNS and known to express ETBRs; hence the researchers we hypothesized that sovateltide could help treat SCI. Receptor binding studies in mammals, including humans, demonstrated that ETARs and ETBRs are distributed throughout the spinal cord [66], and their signaling play critical roles in the inflammatory response and oxidative stress, which are known to affect the neurological recovery after SCI mediated disruption of blood–spinal cord barriers [67][68].
Furthermore, it has been shown that blocking ETARs could significantly reduce the expression of inflammatory factors, e.g., TNF-α, IL-1β, and IL-6 [69]. On the other hand, using a nonselective inhibitor of ET receptors, bosentan inhibited SCI-induced pain response probably by blocking both ETARs and ETBRs [70]. At the same time, the use of SB209670, an ETARs/ETBRs antagonist, to the lesion site of SCI showed the effect on reducing axonal damage after injury [71]. These reports have provided evidence that ETARs, ETBRs, and endogenous ETs play an important role in SCI pathophysiology. Therefore, they could be potential therapeutic targets for the regenerative drug development for SCI to alleviate the effect of primary and secondary injuries, which may ultimately help cure SCI.
Although these studies have shown the involvement of the ET system in SCI, they were based on pharmacologically stimulating or blocking of ETARs or ETARs/ETBRs, and none examined the effect of selectively stimulating ETBRs after SCI. ThWe researchers aand others have shown the role of ETBRs in reducing oxidative stress, increasing blood flow and angiogenesis, enhancing the proliferation of neuronal progenitors and protecting cerebral cortical neurons against apoptosis [17][72][73]. Moreover, theseour findings have shown neuroregeneration and functional recovery following selective ETBRs stimulation by sovateltide in animal models of cerebral ischemia and Alzheimer’s disease [62][33][34]. Since most of the events and factors are common in cerebral and spinal cord injury, such as oxidative stress, inflammation, and neuronal cell death are known to play a critical role in both cases. Like cerebral neuronal regeneration, the generation and proliferation of spinal cord neurons are also essential for functional recovery of the spinal cord after injury. Therefore, thwe researchers hhypothesized that stimulation of ETBRs using sovateltide in the spinal cord would be useful in alleviating the damaging effects and promoting neuronal regeneration and repair after SCI.
4. Neonatal Hypoxic-Ischemic Encephalopathy
Hypoxic-ischemic encephalopathy (HIE) is a major cause of neurologic disabilities and mortality in term neonates, which is mainly caused due to intrapartum complications. Its clinical outcomes are devastating and include neurological disabilities such as cerebral palsy, seizures, and neurodevelopmental disorders in neonates. The incidence of HIE is estimated to be ranged from 1 to 8 per 1000 live births in developed countries and about 26 per 1000 live births in underdeveloped countries [74], constitutes the second cause of mortality in neonates and the third cause of mortality in children < 5 years old [75][76]. The pathophysiology of HIE involves a decrease in placental perfusion or disruption of the delivery of oxygen and glucose in the umbilical cord because of a variety of conditions, including placental abruption, prolapse of the umbilical cord, and uterine rupture. The inadequate placental perfusion causes hypoxia and disrupts the homeostasis in the fetus. Hypoxia leads to a decrease in cardiac output and reduces cerebral blood flow in the fetus. In a moderate decrease, the cerebral blood flow is shunted from the anterior circulation to the posterior circulation, and perfusion in the brainstem, cerebellum, and basal ganglia is maintained. Consequently, the damage is restricted mainly to the cerebral cortex and watershed areas of the cerebral hemispheres. While on the other hand, an acute hypoxia condition invokes an abrupt decrease in cerebral blood flow and produces injury also in the basal ganglia and thalami [77]. The acute hypoxic damage in HIE has been categorized in different phases based on the temporal sequence of the injury. The reduced delivery of oxygen and glucose leads to anaerobic metabolism in the brain, which causes decreased ATP and increased lactic acid production. Due to ATP shortage, transcellular transport is reduced, which causes intracellular accumulation of sodium, water, and calcium. Moreover, upon membrane depolarization, cells release the excitatory amino acid glutamate, and more calcium flows into the cell via N-methyl-D-aspartate–gated channels. The high calcium level in cytosol induces an array of deleterious effects, including necrosis or calpain activation followed by apoptosis, also known as excitotoxicity. Production of free radicals is known to be increased in the hypoxic condition, which causes peroxidation of free fatty acids, which causes more cellular damage. In trauma, neural cells are also known to produce several-fold higher levels of NO, which causes further cellular damage. In summary, the culmination of energy failure, acidosis, glutamate release, lipid peroxidation, and the toxic effect of nitric oxide leads to cell death via necrosis and activates apoptotic cascades in the spinal cord after injury. Depending on the timing of the injury and the available medical intervention, partial recovery may occur during the first 30 to 60 min after the insult. This partial recovery provides a latent phase of injury, which could last from 1 to 6 h and is characterized by recovery of oxidative metabolism, inflammation, and continuation of the activated apoptotic cascades [78]. The secondary phase of injury commonly follows the latent phase within approximately 6 to 15 h after the acute insult. Events in this phase include cytotoxic edema, excitotoxicity, and severe mitochondrial activity failure, leading to cell death and clinical deterioration. The occurrence of seizures is very common in this phase of injury. In some months after the acute insult, a tertiary phase occurs, which involves late cell death, remodeling of the injured brain, and astrogliosis [79]. Neonates with suspected HIE are classified according to the Sarnat staging system [80], which evaluates the level of consciousness, muscle tone, tendon reflexes, complex reflexes, and autonomic function. The Sarnat stage classifies neonatal HIE into the following three categories: stage I (mild), stage II (moderate), and stage III (severe). The pathophysiology of HIE is highly complex, which makes the discovery of effective drugs and interventions very difficult, as evidenced by the failure of several drug trials [81]. At present, there is only one therapy for HIE; therapeutic hypothermia, which has been accepted in its clinical management. The effectiveness of hypothermia in reducing neurological injury caused by HIE has been demonstrated through several clinical trials, including the ICE study [82], Cool Cap study [83], NICHD study [84], and TOBY study [79]. Although the procedure of therapeutic hypothermia is effective, it suffers various limitations, such as the unavailability of trained personnel, equipment, and pediatric neurology support at most of the NICUs (newborn ICUs). Furthermore, its efficacy in preventing neurological disorders in HIE neonates is poor as more than 40% of neonates undergoing hypothermia still develop adverse neurological outcomes [85]. Moreover, the long-term (>2 years) impact of this therapy on neurodevelopment in children remains unclear [86]. Presently, it is a matter of the most concern that a significant number of HIE-affected infants still die or suffer from neurological disabilities whether they receive the hypothermia treatment or not [83][87][88][89]. A decade ago, to address these concerns, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) had invited a panel of experts to review the available evidence, identify knowledge gaps, and suggest research priorities. The panel recommended the development of adjuvant therapies to hypothermia, the use of biomarkers, as well as further refinements in therapeutic hypothermia [88]. Hence, HIE has been an area of global health concern, and the development of new drugs/interventions or adjuvants to therapeutic hypothermia is urgently required. Nonetheless, several adjuvant therapies to hypothermia are undergoing and evaluating the improved survival and neurodevelopmental outcomes in newborns with HIE; more research aiming the treatment optimization and prevention and/or eventually reversal of the HIE damage will be useful. One such potential adjuvant or alternative therapy for neonatal HIE could be a selective agonist of ETBRs such as sovateltide. As previously mentioned, the ouresearch group has demonstrated the role of sovateltide in both neuroprotection and neuroregeneration in adult rat models of cerebral ischemia [29][30]. Additionally, it was found to reduce oxidative stress, increase pro-angiogenic, pro-survival, and anti-apoptotic markers, and increase the number of proliferating cells in rodent brains [28][29][30][31]. Importantly, the ouresearchers' recent studies have shown the role of sovateltide mediated ETBRs stimulation on regulating the mitochondria-mediated apoptotic pathway in rat brains after ischemic stroke damage and its correlation with improved neurological and motor functions in these rats [31]. Furthermore, thwe researchers observed that it improved mitochondrial fusion, decreased fission, increased size, and biogenesis in sovateltide treated rat brains with ischemic stroke [33]. ThWe researchers also demonstrated a novel role of sovateltide in NPCs-mediated regeneration and repair in the rat brain tissues [34]. Therapeutic hypothermia is known to preserve mitochondrial function; it is conceivable that its future adjuvants targeting mitochondria would ensure further efficacy. Hence, it would therefore be of interest to determine the efficacy of sovateltide (which has shown its roles in mitochondrial fate determination as well as in regeneration and repair of neural tissues) in an animal model of neonatal HIE.
References
- Donkor, E.S. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165.
- Bamford, J.; Sandercock, P.; Dennis, M.; Burn, J.; Warlow, C. Classification and natural history of clinically identifiable subtypes of cerebral infarction. Lancet 1991, 337, 1521–1526.
- Seet, R.C.S.; Rabinstein, A.A. Symptomatic Intracranial Hemorrhage following Intravenous Thrombolysis for Acute Ischemic Stroke: A Critical Review of Case Definitions. Cerebrovasc. Dis. 2012, 34, 106–114.
- Minnerup, J.; Wersching, H.; Schilling, M.; Schabitz, W.R. Analysis of early phase and subsequent phase III stroke studies of neuroprotectants: Outcomes and predictors for success. Exp. Transl. Stroke Med. 2014, 6, 2.
- Gulati, A.; Hornick, M.G.; Briyal, S.; Lavhale, M.S. A novel neuroregenerative approach using ET(B) receptor agonist, IRL-1620, to treat CNS disorders. Physiol. Res. 2018, 67, S95–S113.
- Lampl, Y.; Fleminger, G.; Gilad, R.; Galron, R.; Sarova-Pinhas, I.; Sokolovsky, M. Endothelin in cerebrospinal fluid and plasma of patients in the early stage of ischemic stroke. Stroke J. Cereb. Circ. 1997, 28, 1951–1955.
- Ziv, I.; Fleminger, G.; Djaldetti, R.; Achiron, A.; Melamed, E.; Sokolovsky, M. Increased plasma endothelin-1 in acute ischemic stroke. Stroke J. Cereb. Circ. 1992, 23, 1014–1016.
- Barone, F.C.; Ohlstein, E.H.; Hunter, A.J.; Campbell, C.A.; Hadingham, S.H.; Parsons, A.A.; Yang, Y.; Shohami, E. Selective antagonism of endothelin-A-receptors improves outcome in both head trauma and focal stroke in rat. J. Cardiovasc. Pharmacol. 2000, 36, S357–S361.
- Briyal, S.; Gulati, A. Endothelin-A receptor antagonist BQ123 potentiates acetaminophen induced hypothermia and reduces infarction following focal cerebral ischemia in rats. Eur. J. Pharmacol. 2010, 644, 73–79.
- Legos, J.J.; Lenhard, S.C.; Haimbach, R.E.; Schaeffer, T.R.; Bentley, R.G.; McVey, M.J.; Chandra, S.; Irving, E.A.; Andrew, A.P.; Barone, F.C. SB 234551 selective ET(A) receptor antagonism: Perfusion/diffusion MRI used to define treatable stroke model, time to treatment and mechanism of protection. Exp. Neurol. 2008, 212, 53–62.
- Tatlisumak, T.; Carano, R.A.; Takano, K.; Opgenorth, T.J.; Sotak, C.H.; Fisher, M. A novel endothelin antagonist, A-127722, attenuates ischemic lesion size in rats with temporary middle cerebral artery occlusion: A diffusion and perfusion MRI study. Stroke J. Cereb. Circ. 1998, 29, 850–857; discussion 857–858.
- Zhang, R.L.; Zhang, C.; Zhang, L.; Roberts, C.; Lu, M.; Kapke, A.; Cui, Y.; Ninomiya, M.; Nagafuji, T.; Albala, B.; et al. Synergistic effect of an endothelin type A receptor antagonist, S-0139, with rtPA on the neuroprotection after embolic stroke. Stroke J. Cereb. Circ. 2008, 39, 2830–2836.
- Briyal, S.; Gulati, A.; Gupta, Y.K. Effect of combination of endothelin receptor antagonist (TAK-044) and aspirin in middle cerebral artery occlusion model of acute ischemic stroke in rats. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 257–263.
- Briyal, S.; Pant, A.B.; Gupta, Y.K. Protective effect of endothelin antagonist (TAK-044) on neuronal cell viability in in vitro oxygen-glucose deprivation model of stroke. Indian J. Physiol. Pharmacol. 2006, 50, 157–162.
- Chuquet, J.; Benchenane, K.; Toutain, J.; MacKenzie, E.T.; Roussel, S.; Touzani, O. Selective blockade of endothelin-B receptors exacerbates ischemic brain damage in the rat. Stroke J. Cereb. Circ. 2002, 33, 3019–3025.
- Ehrenreich, H.; Oldenburg, J.; Hasselblatt, M.; Herms, J.; Dembowski, C.; Loffler, B.M.; Bruck, W.; Kamrowski-Kruck, H.; Gall, S.; Siren, A.L.; et al. Endothelin B receptor-deficient rats as a subtraction model to study the cerebral endothelin system. Neuroscience 1999, 91, 1067–1075.
- Ehrenreich, H.; Nau, T.R.; Dembowski, C.; Hasselblatt, M.; Barth, M.; Hahn, A.; Schilling, L.; Siren, A.L.; Bruck, W. Endothelin b receptor deficiency is associated with an increased rate of neuronal apoptosis in the dentate gyrus. Neuroscience 2000, 95, 993–1001.
- Castaneda, M.M.; Cubilla, M.A.; Lopez-Vicchi, M.M.; Suburo, A.M. Endothelinergic cells in the subependymal region of mice. Brain Res. 2010, 1321, 20–30.
- Gulati, A.; Kumar, A.; Morrison, S.; Shahani, B.T. Effect of centrally administered endothelin agonists on systemic and regional blood circulation in the rat: Role of sympathetic nervous system. Neuropeptides 1997, 31, 301–309.
- Koyama, Y.; Takemura, M.; Fujiki, K.; Ishikawa, N.; Shigenaga, Y.; Baba, A. BQ788, an endothelin ET(B) receptor antagonist, attenuates stab wound injury-induced reactive astrocytes in rat brain. Glia 1999, 26, 268–271.
- Sakurai-Yamashita, Y.; Niwa, M.; Yamashita, K.; Kataoka, Y.; Himeno, A.; Shigematsu, K.; Tsutsumi, K.; Taniyama, K. Endothelin receptors in kainic acid-induced neural lesions of rat brain. Neuroscience 1997, 81, 565–577.
- Yamashita, K.; Niwa, M.; Kataoka, Y.; Shigematsu, K.; Himeno, A.; Tsutsumi, K.; Nakano-Nakashima, M.; Sakurai-Yamashita, Y.; Shibata, S.; Taniyama, K. Microglia with an endothelin ETB receptor aggregate in rat hippocampus CA1 subfields following transient forebrain ischemia. J. Neurochem. 1994, 63, 1042–1051.
- Vidovic, M.; Chen, M.M.; Lu, Q.Y.; Kalloniatis, K.F.; Martin, B.M.; Tan, A.H.; Lynch, C.; Croaker, G.D.; Cass, D.T.; Song, Z.M. Deficiency in endothelin receptor B reduces proliferation of neuronal progenitors and increases apoptosis in postnatal rat cerebellum. Cell. Mol. Neurobiol. 2008, 28, 1129–1138.
- Siren, A.L.; Lewczuk, P.; Hasselblatt, M.; Dembowski, C.; Schilling, L.; Ehrenreich, H. Endothelin B receptor deficiency augments neuronal damage upon exposure to hypoxia-ischemia in vivo. Brain Res. 2002, 945, 144–149.
- Leonard, M.G.; Prazad, P.; Puppala, B.; Gulati, A. Selective Endothelin-B Receptor Stimulation Increases Vascular Endothelial Growth Factor in the Rat Brain during Postnatal Development. Drug Res. 2015, 65, 607–613.
- Puppala, B.; Awan, I.; Briyal, S.; Mbachu, O.; Leonard, M.; Gulati, A. Ontogeny of endothelin receptors in the brain, heart, and kidneys of neonatal rats. Brain Dev. 2015, 37, 206–215.
- Gulati, A. Endothelin Receptors, Mitochondria and Neurogenesis in Cerebral Ischemia. Curr. Neuropharmacol. 2016, 14, 619–626.
- Leonard, M.G.; Briyal, S.; Gulati, A. Endothelin B receptor agonist, IRL-1620, reduces neurological damage following permanent middle cerebral artery occlusion in rats. Brain Res. 2011, 1420, 48–58.
- Leonard, M.G.; Briyal, S.; Gulati, A. Endothelin B receptor agonist, IRL-1620, provides long-term neuroprotection in cerebral ischemia in rats. Brain Res. 2012, 1464, 14–23.
- Leonard, M.G.; Gulati, A. Endothelin B receptor agonist, IRL-1620, enhances angiogenesis and neurogenesis following cerebral ischemia in rats. Brain Res. 2013, 1528, 28–41.
- Briyal, S.; Ranjan, A.K.; Hornick, M.G.; Puppala, A.K.; Luu, T.; Gulati, A. Anti-apoptotic activity of ETB receptor agonist, IRL-1620, protects neural cells in rats with cerebral ischemia. Sci. Rep. 2019, 9, 10439.
- Cifuentes, E.G.; Hornick, M.G.; Havalad, S.; Donovan, R.L.; Gulati, A. Neuroprotective Effect of IRL-1620, an Endothelin B Receptor Agonist, on a Pediatric Rat Model of Middle Cerebral Artery Occlusion. Front. Pediatr. 2018, 6, 310.
- Ranjan, A.K.; Briyal, S.; Gulati, A. Sovateltide (IRL-1620) activates neuronal differentiation and prevents mitochondrial dysfunction in adult mammalian brains following stroke. Sci. Rep. 2020, 10, 12737.
- Ranjan, A.K.; Briyal, S.; Khandekar, D.; Gulati, A. Sovateltide (IRL-1620) affects neuronal progenitors and prevents cerebral tissue damage after ischemic stroke. Can. J. Physiol. Pharmacol. 2020, 98, 659–666.
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020.
- Crous-Bou, M.; Minguillon, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimer’s Res. Ther. 2017, 9, 71.
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res 2018, 7, 1161.
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031.
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220.
- Cummings, J.L.; Ringman, J.; Vinters, H.V. Neuropathologic correlates of trial-related instruments for Alzheimer’s disease. Am. J. Neurodegener. Dis. 2014, 3, 45–49.
- Emanuel, E.J. A Middle Ground for Accelerated Drug Approval-Lessons from Aducanumab. JAMA 2021, 326, 1367–1368.
- Dunn, B.; Stein, P.; Temple, R.; Cavazzoni, P. An Appropriate Use of Accelerated Approval—Aducanumab for Alzheimer’s Disease. N. Engl. J. Med. 2021, 385, 856–857.
- Alexander, G.C.; Knopman, D.S.; Emerson, S.S.; Ovbiagele, B.; Kryscio, R.J.; Perlmutter, J.S.; Kesselheim, A.S. Revisiting FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 769–771.
- Gulati, A.; Agrawal, N.; Vibha, D.; Misra, U.K.; Paul, B.; Jain, D.; Pandian, J.; Borgohain, R. Safety and Efficacy of Sovateltide (IRL-1620) in a Multicenter Randomized Controlled Clinical Trial in Patients with Acute Cerebral Ischemic Stroke. CNS Drugs 2021, 35, 85–104.
- Palmer, J.C.; Baig, S.; Kehoe, P.G.; Love, S. Endothelin-converting enzyme-2 is increased in Alzheimer’s disease and up-regulated by Abeta. Am. J. Pathol. 2009, 175, 262–270.
- Koyama, Y. Endothelin systems in the brain: Involvement in pathophysiological responses of damaged nerve tissues. Biomol. Concepts 2013, 4, 335–347.
- Turner, A.J.; Murphy, L.J. Molecular pharmacology of endothelin converting enzymes. Biochem. Pharmacol. 1996, 51, 91–102.
- Rodriguiz, R.M.; Gadnidze, K.; Ragnauth, A.; Dorr, N.; Yanagisawa, M.; Wetsel, W.C.; Devi, L.A. Animals lacking endothelin-converting enzyme-2 are deficient in learning and memory. Genes Brain Behav. 2008, 7, 418–426.
- Eckman, E.A.; Watson, M.; Marlow, L.; Sambamurti, K.; Eckman, C.B. Alzheimer’s disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. 2003, 278, 2081–2084.
- Yamashita, K.I.; Taniwaki, Y.; Utsunomiya, H.; Taniwaki, T. Cerebral blood flow reduction associated with orientation for time in amnesic mild cognitive impairment and Alzheimer disease patients. J. Neuroimaging 2014, 24, 590–594.
- Gulati, A.; Kumar, A.; Shahani, B.T. Cardiovascular effects of centrally administered endothelin-1 and its relationship to changes in cerebral blood flow. Life Sci. 1996, 58, 437–445.
- Gulati, A.; Rebello, S.; Roy, S.; Saxena, P.R. Cardiovascular effects of centrally administered endothelin-1 in rats. J. Cardiovasc. Pharmacol. 1995, 26 (Suppl. 3), S244–S246.
- Paris, D.; Humphrey, J.; Quadros, A.; Patel, N.; Crescentini, R.; Crawford, F.; Mullan, M. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: Role of inflammation. Neurol. Res. 2003, 25, 642–651.
- Briyal, S.; Philip, T.; Gulati, A. Endothelin-A receptor antagonists prevent amyloid-beta-induced increase in ETA receptor expression, oxidative stress, and cognitive impairment. J. Alzheimer’s Dis. 2011, 23, 491–503.
- Druckenbrod, N.R.; Powers, P.A.; Bartley, C.R.; Walker, J.W.; Epstein, M.L. Targeting of endothelin receptor-B to the neural crest. Genesis 2008, 46, 396–400.
- Nishikawa, K.; Ayukawa, K.; Hara, Y.; Wada, K.; Aoki, S. Endothelin/endothelin-B receptor signals regulate ventricle-directed interkinetic nuclear migration of cerebral cortical neural progenitors. Neurochem. Int. 2011, 58, 261–272.
- Yagami, T.; Ueda, K.; Asakura, K.; Kuroda, T.; Hata, S.; Sakaeda, T.; Kambayashi, Y.; Fujimoto, M. Effects of endothelin B receptor agonists on amyloid beta protein (25–35)-induced neuronal cell death. Brain Res. 2002, 948, 72–81.
- Leonard, M.G.; Gulati, A. Repeated administration of ET(B) receptor agonist, IRL-1620, produces tachyphylaxis only to its hypotensive effect. Pharmacol. Res. 2009, 60, 402–410.
- Joshi, M.D.; Oesterling, B.M.; Wu, C.; Gwizdz, N.; Pais, G.; Briyal, S.; Gulati, A. Evaluation of liposomal nanocarriers loaded with ETB receptor agonist, IRL-1620, using cell-based assays. Neuroscience 2016, 312, 141–152.
- Briyal, S.; Shepard, C.; Gulati, A. Endothelin receptor type B agonist, IRL-1620, prevents beta amyloid (Abeta) induced oxidative stress and cognitive impairment in normal and diabetic rats. Pharmacol. Biochem. Behav. 2014, 120, 65–72.
- Briyal, S.; Nguyen, C.; Leonard, M.; Gulati, A. Stimulation of endothelin B receptors by IRL-1620 decreases the progression of Alzheimer’s disease. Neuroscience 2015, 301, 1–11.
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic Spinal Cord Injury: An Overview of Pathophysiology, Models and Acute Injury Mechanisms. Front. Neurol. 2019, 10, 282.
- Nori, S.; Ahuja, C.S.; Fehlings, M.G. Translational Advances in the Management of Acute Spinal Cord Injury: What is New? What is Hot? Neurosurgery 2017, 64, 119–128.
- Donovan, J.; Kirshblum, S. Clinical Trials in Traumatic Spinal Cord Injury. Neurotherapeutics 2018, 15, 654–668.
- Peters, C.M.; Rogers, S.D.; Pomonis, J.D.; Egnaczyk, G.F.; Keyser, C.P.; Schmidt, J.A.; Ghilardi, J.R.; Maggio, J.E.; Mantyh, P.W. Endothelin receptor expression in the normal and injured spinal cord: Potential involvement in injury-induced ischemia and gliosis. Exp. Neurol. 2003, 180, 1–13.
- Kallakuri, S.; Kreipke, C.W.; Schafer, P.C.; Schafer, S.M.; Rafols, J.A. Brain cellular localization of endothelin receptors A and B in a rodent model of diffuse traumatic brain injury. Neuroscience 2010, 168, 820–830.
- McKenzie, A.L.; Hall, J.J.; Aihara, N.; Fukuda, K.; Noble, L.J. Immunolocalization of endothelin in the traumatized spinal cord: Relationship to blood-spinal cord barrier breakdown. J. Neurotrauma 1995, 12, 257–268.
- Guo, J.; Li, Y.; He, Z.; Zhang, B.; Li, Y.; Hu, J.; Han, M.; Xu, Y.; Li, Y.; Gu, J.; et al. Targeting endothelin receptors A and B attenuates the inflammatory response and improves locomotor function following spinal cord injury in mice. Int. J. Mol. Med. 2014, 34, 74–82.
- Forner, S.; Martini, A.C.; Andrade, E.L.; Rae, G.A. Neuropathic pain induced by spinal cord injury: Role of endothelin ETA and ETB receptors. Neurosci. Lett. 2016, 617, 14–21.
- Uesugi, M.; Kasuya, Y.; Hayashi, K.; Goto, K. SB209670, a potent endothelin receptor antagonist, prevents or delays axonal degeneration after spinal cord injury. Brain Res. 1998, 786, 235–239.
- Laziz, I.; Larbi, A.; Grebert, D.; Sautel, M.; Congar, P.; Lacroix, M.C.; Salesse, R.; Meunier, N. Endothelin as a neuroprotective factor in the olfactory epithelium. Neuroscience 2011, 172, 20–29.
- Yagami, T.; Ueda, K.; Sakaeda, T.; Okamura, N.; Nakazato, H.; Kuroda, T.; Hata, S.; Sakaguchi, G.; Itoh, N.; Hashimoto, Y.; et al. Effects of an endothelin B receptor agonist on secretory phospholipase A2-IIA-induced apoptosis in cortical neurons. Neuropharmacology 2005, 48, 291–300.
- Kurinczuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 329–338.
- Lawn, J.E.; Blencowe, H.; Oza, S.; You, D.; Lee, A.C.; Waiswa, P.; Lalli, M.; Bhutta, Z.; Barros, A.J.; Christian, P.; et al. Every Newborn: Progress, priorities, and potential beyond survival. Lancet 2014, 384, 189–205.
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000-15: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016, 388, 3027–3035.
- Harteman, J.C.; Nikkels, P.G.; Benders, M.J.; Kwee, A.; Groenendaal, F.; de Vries, L.S. Placental pathology in full-term infants with hypoxic-ischemic neonatal encephalopathy and association with magnetic resonance imaging pattern of brain injury. J. Pediatr. 2013, 163, 968–995.e2.
- Azzopardi, D.; Wyatt, J.S.; Cady, E.B.; Delpy, D.T.; Baudin, J.; Stewart, A.L.; Hope, P.L.; Hamilton, P.A.; Reynolds, E.O. Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1989, 25, 445–451.
- Azzopardi, D.V.; Strohm, B.; Edwards, A.D.; Dyet, L.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; Levene, M.; Marlow, N.; Porter, E.; et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 2009, 361, 1349–1358.
- Bale, G.; Mitra, S.; de Roever, I.; Sokolska, M.; Price, D.; Bainbridge, A.; Gunny, R.; Uria-Avellanal, C.; Kendall, G.S.; Meek, J.; et al. Oxygen dependency of mitochondrial metabolism indicates outcome of newborn brain injury. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2019, 39, 2035–2047.
- Yildiz, E.P.; Ekici, B.; Tatli, B. Neonatal hypoxic ischemic encephalopathy: An update on disease pathogenesis and treatment. Expert Rev. Neurother. 2017, 17, 449–459.
- Jacobs, S.E.; Morley, C.J.; Inder, T.E.; Stewart, M.J.; Smith, K.R.; McNamara, P.J.; Wright, I.M.; Kirpalani, H.M.; Darlow, B.A.; Doyle, L.W.; et al. Whole-body hypothermia for term and near-term newborns with hypoxic-ischemic encephalopathy: A randomized controlled trial. Arch. Pediatr. Adolesc. Med. 2011, 165, 692–700.
- Gluckman, P.D.; Wyatt, J.S.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Polin, R.A.; Robertson, C.M.; Thoresen, M.; Whitelaw, A.; et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: Multicentre randomised trial. Lancet 2005, 365, 663–670.
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584.
- Edwards, A.D.; Brocklehurst, P.; Gunn, A.J.; Halliday, H.; Juszczak, E.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: Synthesis and meta-analysis of trial data. BMJ 2010, 340, c363.
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N. Engl. J. Med. 2012, 366, 2085–2092.
- Gunn, A.J.; Gluckman, P.D.; Gunn, T.R. Selective head cooling in newborn infants after perinatal asphyxia: A safety study. Pediatrics 1998, 102, 885–892.
- Higgins, R.D.; Raju, T.; Edwards, A.D.; Azzopardi, D.V.; Bose, C.L.; Clark, R.H.; Ferriero, D.M.; Guillet, R.; Gunn, A.J.; Hagberg, H.; et al. Hypothermia and other treatment options for neonatal encephalopathy: An executive summary of the Eunice Kennedy Shriver NICHD workshop. J. Pediatr. 2011, 159, 851–858.e1.
- Thoresen, M.; Whitelaw, A. Cardiovascular changes during mild therapeutic hypothermia and rewarming in infants with hypoxic-ischemic encephalopathy. Pediatrics 2000, 106, 92–99.
More