Cellular redox homeoHstasis is precisely balanced by generation and elimination of reactive oxygen species (ROS). ROS are not only capable of causing oxidation of proteins, lipids and DNA to damage cells but can also act as signaling molecules to modulate transcription factors and epigenetic pathways that determine cell survival and death. Hsp70 proteins are central hubs for p70 is the key chaperone in protein quality control and the central hub of the cellular proteostasis and are important factors to ameliorate damage from different kinds of stress including oxidative stress. Hsp70 members often participate in different cellular signaling pathways via their clients and cochaperones. ROS can directly cause oxidative cysteine modifications of network, participating in numerous cellular processes by interacting with different clients. Hsp70 members to alter their structure and chaperone activity, resulting in changes in the interactions between Hsp70 and their clients or cochaperones, which can then transfer redox signals to Hsp70-related signaling pathways. On the other hand, ROS also activate some redox-related signaling pathways to indirectly modulateis tightly related to redox homeostasis in several ways, including functional regulation of Hsp70 activity and expression. Pocaused by post-translational modifications including phosphorylation together with elevated Hsp70 (especially cysteine modifications), induced expression can expand the capacity n of Hsp70 to deal with ROS-damaged proteins and support antioxidant enzymes. Knowledge about the response and role of Hsp70 in redox homcaused by oxidative stress, Hsp70-dependent proteostasis will facilitate our understanding of the cellular knock-on effects of inhibitors targeting Hsp70 and the mechanisms of under oxidative stress and redox-related diseases and agingsignaling pathways involving Hsp70.
- redox homeostasis
- oxidative stress
- ROS
- Hsp70
- cysteine modifications
- glutathionylation
1. Introduction
| Cellular Processes | Critical Signaling Molecules Modified by ROS |
|---|---|
| proliferation and survival | mitogen-activated protein kinases (MAPKs), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), phosphatase and tensin homolog deleted on chromosome ten (PTEN) and protein tyrosine phosphatases |
| redox homeostasis | thioredoxin, peroxiredoxin, redox factor-1 (Ref-1) and Kelch-like ECH-associated protein 1 (Keap1)/nuclear factor erythroid 2-related factor 2 (Nrf2) |
| mitochondrial oxidative stress and aging | p66Shc |
| iron homeostasis | iron response element–iron regulatory protein (IRE-IRP) containing iron–sulfur cluster |
| DNA damage response | ataxia telangiectasia mutated (ATM) |
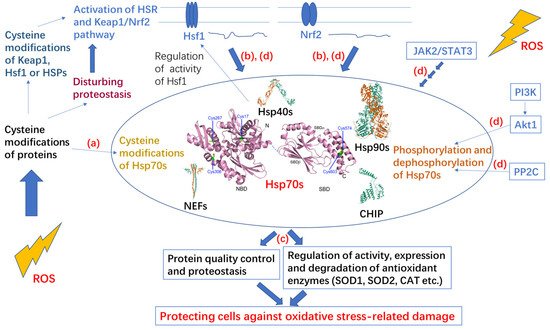
2. Hsp70 System
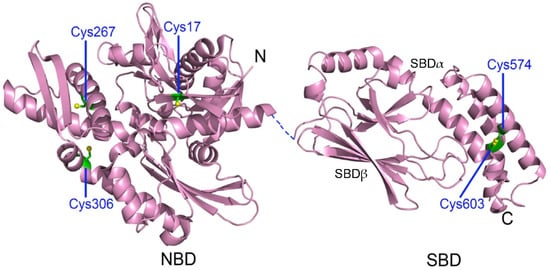
3. Post-Translational Modifications of Hsp70 under Oxidative Stress
3.1. Cysteine Oxidation of Hsp70 and Redox Homeostasis
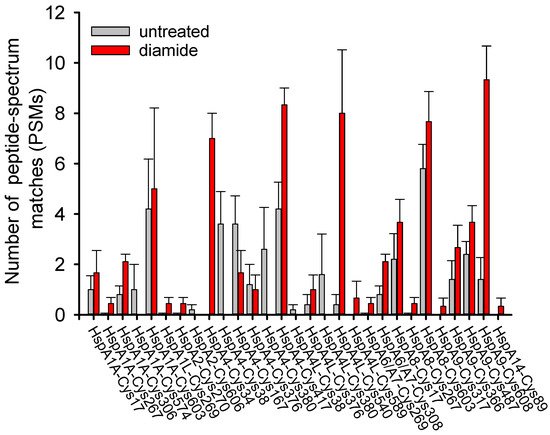

3.2. Covalent Modifications of Hsp70 and Redox Homeostasis
4. Protection Effect and Upregulated Expression of Hsp70 under Oxidative Stress
Cellular redox homeostasis is precisely balanced by generation and elimination of reactive oxygen species (ROS). ROS are not only capable of causing oxidation of proteins, lipids and DNA to damage cells but can also act as signaling molecules to modulate transcription factors and epigenetic pathways that determine cell survival and death. Hsp70 proteins are central hubs for proteostasis and are important factors to ameliorate damage from different kinds of stress including oxidative stress. Hsp70 members often participate in different cellular signaling pathways via their clients and cochaperones. ROS can directly cause oxidative cysteine modifications of Hsp70 members to alter their structure and chaperone activity, resulting in changes in the interactions between Hsp70 and their clients or cochaperones, which can then transfer redox signals to Hsp70-related signaling pathways. On the other hand, ROS also activate some redox-related signaling pathways to indirectly modulate Hsp70 activity and expression. Post-translational modifications including phosphorylation together with elevated Hsp70 expression can expand the capacity of Hsp70 to deal with ROS-damaged proteins and support antioxidant enzymes. Knowledge about the response and role of Hsp70 in redox homeostasis will facilitate our understanding of the cellular knock-on effects of inhibitors targeting Hsp70 and the mechanisms of redox-related diseases and aging.
Hsp70 can counteract oxidative stress damage to proteins by inhibiting aggregation and/or facilitating degradation of oxidized proteins, as well as inducing the expression of key antioxidant enzymes such as SOD1 and CAT and adjusting the activity of antioxidant enzymes [15, 89-92]. Overexpression of HSP70 in the muscle of mice antagonizes the aging-related increase in protein cysteine modifications including carbonylation, oxidation and formation of disulfide bonds [93]. Overexpression of mitochondrial HSP70/Hsp75 in rat brain protects mitochondria and results in the marked reduction in free radical generation [94]. Under oxidative stress, HspA1A and HspA8 can mediate the C-terminus of Hsp70-interacting protein (CHIP, also known as STUB1) to recognize and ubiquitinate ROS-stressed peroxisomes, resulting in their turnover by autophagy [95]. Overexpression of HSP70 suppresses ROS production from mitochondria in human lung microvascular endothelial cells (HLMVEC) exposed to bacterial toxins [96]. The HSPA1B rs1061581 polymorphism (1267 A/G), which is linked to the risk of developing multiple sclerosis, is related to ROS levels and has a role in the variation in HSPA1B expression levels under oxidative stimulus [97].Hsp70 can buffer different kinds of stress including oxidative stress to contribute to redox homeostasis. Hsp70 members are important antioxidative components in eliminating damaged oxidized proteins and support antioxidative enzymes. Under oxidative stress, Hsp70 members will inevitably undergo oxidization, resulting in changes in their structure and chaperone activity. However, cysteine modifications often downregulate the chaperone activity of Hsp70, which is disadvantageous for dealing with protein damage due to oxidation. From this point of view, cysteine residues in Hsp70 are not conducive towards Hsp70 exerting its antioxidative functions. At the same time, only very few Hsp70 members do not contain any cysteine residues, and the number of cysteine residues in Hsp70 increases with evolution, suggesting important roles of cysteine residues in Hsp70. Hsp70 members can sense redox by cysteine modifications, then transfer redox signals by modulating the interaction between Hsp70 members and their clients. Hsp70 members are involved in cellular signaling due to the fact that their clients include key molecules in signaling pathways, including Hsf1 and Akt1. Cochaperones also guide Hsp70 members within different cellular processes, such as CHIP, acting as a link between Hsp70 and the ubiquitin–proteasome system (UPS) for degradation of Hsp70 clients. Thus, cysteine modifications of Hsp70 also change the fate of the clients by modifying the cooperation between Hsp70 and cochaperones. Therefore, cysteine modifications of Hsp70 may act to amplify the transfer of redox signaling to a broader range of proteins, achieving greater antioxidative potential in a shorter time and restoring the cellular environment by multiple pathways. As a consequence, Hsp70 proteins are expressed much more rapidly through different signaling pathways, and the newly synthesized Hsp70 proteins are not oxidized and have normal chaperone activity to deal with the damaged proteins that have accumulated during oxidative stress. Thus, combining an in-depth understanding of the basic principles of Hsp70 systems (including the relationship between the structure and function of Hsp70, the interaction between Hsp70 and its cochaperones, and the interaction between Hsp70 and its clients) and the extensive exploration of redox-related signaling pathways involving Hsp70 will lead to a fuller understanding of Hsp70 function in redox homeostasis.
Oxidative stress can activate the HSR and Kelch-like ECH-associated protein 1 (Keap1)/Nrf2 pathway to upregulate expression of HSP70, and the increased protein level of Hsp70 protects cells against oxidative-stress-related damage [15, 98, 99]. However, different types of oxidative stress differ in their capacity to induce the HSR and elevate expression of HSP70, and the same oxidative stimulus may lead to distinct stress responses in different cells [100-102]. Oxidative-stress induced by 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) enhances HSP70 expression to exert anti-inflammatory effects [103]. Under mild oxidative stress conditions, expression of HSPA1A/B is upregulated in the mammalian neuronal cell line HT22 [90, 104]. In the early phase after oxidative stress, HspA1A/B binds to partially unfolded oxidized proteins to prevent their aggregation [104]. Then, HspA1A/B facilitates the degradation of bound oxidized proteins by 20S proteasomes [104]. Expression of HspA8 is not affected by oxidative stress, and HspA8 has no effect on removal of proteins damaged by oxidation by the 20S proteasome [104]. Thus, HspA1A/B but not HspA8 mediates degradation of damaged proteins by the UPS under oxidative stress conditions [104], while HspA8-facilitated CMA is also activated by oxidative stress to facilitate degradation of oxidized proteins [105, 106].
5. Hsp70 Participates in Multiple Redox-Related Signaling Pathways
Hsp70 and redox homeostasis are linked by multiple signaling pathways including inositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (PKB, Akt)/Hsp70, Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3)/Hsp70, and Keap1/Nrf2/Hsf1/Hsp70. Expression or activity of Hsp70 is often modulated by the related signaling pathways [107-109].
SOD2 protects cells against mitochondrial oxidative damage [110]. HspA1A can bind to SOD2 to prevent its aggregation and control its CHIP-mediated degradation or import it into mitochondria [107, 111]. CHIP cooperates with HspA1A by binding to the C-terminus EEVD motif and the SBDα of HspA1A [112]. PI3K/Akt signaling is crucial for receiving input from the cell membrane and mitochondria [113]. Akt1 is the downstream kinase of PI3K and a client of HspA1A. Akt1 binds to the SBDα of HspA1A and phosphorylates the C-terminus Ser631 of HspA1A, resulting in decoupling of HspA1A and CHIP and inhibition of the degradation of SOD2 [107]. Then, the import of SOD2 into mitochondria is promoted. As a negative feedback loop, the resulting decrease in oxidative stress activation permits phosphatase 2C (PP2C) to dephosphorylate HspA1A to promote degradation of SOD2 [107]. Thus, Hsp70 phosphorylation involved in the PI3K/Akt/Hsp70 signaling pathway contributes to redox homeostasis.
The oncogenic signaling pathway JAK2/STAT3 plays crucial roles in regulating apoptosis, proliferation, differentiation, and the inflammatory response and participates in the occurrence and development of various tumors [114]. AG490 is an inhibitor of the JAK2/STAT3 signaling pathway [115]. It was found that H2O2-induced time-dependent increase in Hsp70 protein expression in vascular smooth muscle cells is inhibited by pretreatment with AG-490, suggesting that ROS can regulate HSP70 expression via the JAK2/STAT3 signaling pathway [116]. It was also found that both HSP70 RNAi and AG490 can increase ROS levels in Burkitt’s lymphoma Raji cells and reduce the activity of SOD and GPX, suggesting that inhibiting Hsp70 or the JAK2/STAT3 pathway may induce oxidative stress of Raji cells [108]. Therefore, the JAK2/STAT3 signaling pathway may contribute to redox homeostasis by modulating the expression of HSP70.
The Keap1/Nrf2 pathway is a thiol-based sensor–effector for maintaining redox homeostasis [16]. Keap1 acts as a cysteine-thiol-rich sensor of redox status, and Nrf2 is a transcription factor for some cytoprotective genes including some enzymatic antioxidants and Hsp70 [16]. The Keap1/Nrf2 complex facilitates degradation of Nrf2 and represses the activity of Nrf2 [16]. ROS or electrophiles can cause cysteine modification of Keap1, which contains multiple active thiols, to weaken the interaction between Keap1 and Nrf2 and promote the entrance of Nrf2 into the cell nucleus and activation of Nrf2 [16, 117]. Hsf1 is an important transcription factor for genes related to HSR. Hsp90, Hsp70 and Hsp40 cooperate to inactivate Hsf1 by binding to it and preventing it from entering the cell nucleus, and they are also transcription targets of Hsf1, forming a negative feedback regulatory loop [118, 119]. Although the mechanism by which Hsf1 is activated has not been fully elucidated, activation of Hsf1 is thought to be related to stress damage of proteins and modification of HSPs or Hsf1 [117, 120]. Both Nrf2 and Hsf1 are critical for adaptation and survival, and there is crosstalk between the two pathways through overlapping transcriptional targets including HSP70 [99]. Some reports indicate that the Keap1/Nrf2 pathway is activated before the heat shock response upon oxidative or electrophilic stress [99, 121, 122]. When Hsf1 is mutated and therefore unable to activate the HSR, Nrf2 can be activated by heat shock to cause delayed upregulation of HSP70, suggesting the two pathways may compensate for each other [123]. Oxidative stress resulting from a lack of methionine induces HSPA1A expression through Nrf2 activation but not Hsf1 activation in HEK293 cells [109]. Both Hsf1 and Nrf2 pathways contribute to redox homeostasis and cooperate to promote a more reducing cellular environment.
6. Conclusions and Perspectives
Redox homeostasis relies on the balance between ROS generation and the antioxidation system. Hsp70 can buffer different kinds of stress including oxidative stress. Hsp70 members are important antioxidative components in eliminating damaged oxidized proteins and support antioxidative enzymes. Under oxidative stress, Hsp70 members will inevitably undergo oxidization, resulting in changes in their structure and chaperone activity. However, cysteine modifications often downregulate the chaperone activity of Hsp70, which is disadvantageous for dealing with protein damage due to oxidation. From this point of view, cysteine residues in Hsp70 are not conducive towards Hsp70 exerting its antioxidative functions. At the same time, only very few Hsp70 members do not contain any cysteine residues, and the number of cysteine residues in Hsp70 increases with evolution, suggesting important roles of cysteine residues in Hsp70. Hsp70 members can sense redox by cysteine modifications, then transfer redox signals by modulating the interaction between Hsp70 members and their clients. Hsp70 members are involved in cellular signaling due to the fact that their clients include key molecules in signaling pathways, including Hsf1 and Akt1. Cochaperones also guide Hsp70 members within different cellular processes, such as CHIP, acting as a link between Hsp70 and the UPS for degradation of Hsp70 clients. Thus, cysteine modifications of Hsp70 also change the fate of the clients by modifying the cooperation between Hsp70 and cochaperones. Therefore, cysteine modifications of Hsp70 may act to amplify the transfer of redox signaling to a broader range of proteins, achieving greater antioxidative potential in a shorter time and restoring the cellular environment by multiple pathways. As a consequence, Hsp70 proteins are expressed much more rapidly through different signaling pathways, and the newly synthesized Hsp70 proteins are not oxidized and have normal chaperone activity to deal with the damaged proteins that have accumulated during oxidative stress. Thus, combining an in-depth understanding of the basic principles of Hsp70 systems (including the relationship between the structure and function of Hsp70, the interaction between Hsp70 and its cochaperones, and the interaction between Hsp70 and its clients) and the extensive exploration of redox-related signaling pathways involving Hsp70 will lead to a fuller understanding of Hsp70 function in redox homeostasis.References
- Holmström, K.M. and Finkel, T., Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol, 2014, 15(6), 411-21, DOI: 10.1038/nrm3801.
- Kong, H. and Chandel, N.S., Regulation of redox balance in cancer and T cells. J Biol Chem, 2018, 293(20), 7499-7507, DOI: 10.1074/jbc.TM117.000257.
- Zhang, L., Wang, X., Cueto, R., Effi, C., Zhang, Y., Tan, H., Qin, X., Ji, Y., Yang, X., and Wang, H., Biochemical basis and metabolic interplay of redox regulation. Redox Biol, 2019, 26, 101284, DOI: 10.1016/j.redox.2019.101284.
- Sies, H., Oxidative stress: a concept in redox biology and medicine. Redox Biol, 2015, 4, 180-3, DOI: 10.1016/j.redox.2015.01.002.
- Ray, P.D., Huang, B.W., and Tsuji, Y., Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal, 2012, 24(5), 981-90, DOI: 10.1016/j.cellsig.2012.01.008.
- Tabak, O., Gelisgen, R., Erman, H., Erdenen, F., Muderrisoglu, C., Aral, H., and Uzun, H., Oxidative lipid, protein, and DNA damage as oxidative stress markers in vascular complications of diabetes mellitus. Clin Invest Med, 2011, 34(3), E163-71, DOI: 10.25011/cim.v34i3.15189.
- Liu, J., Wang, X., Shigenaga, M.K., Yeo, H.C., Mori, A., and Ames, B.N., Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. Faseb j, 1996, 10(13), 1532-8.
- Yang, W.S. and Stockwell, B.R., Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol, 2016, 26(3), 165-176, DOI: 10.1016/j.tcb.2015.10.014.
- Forman, H.J., Ursini, F., and Maiorino, M., An overview of mechanisms of redox signaling. J Mol Cell Cardiol, 2014, 73, 2-9, DOI: 10.1016/j.yjmcc.2014.01.018.
- Forman, H.J., Redox signaling: An evolution from free radicals to aging. Free Radic Biol Med, 2016, 97, 398-407, DOI: 10.1016/j.freeradbiomed.2016.07.003.
- Turell, L., Zeida, A., and Trujillo, M., Mechanisms and consequences of protein cysteine oxidation: the role of the initial short-lived intermediates. Essays Biochem, 2020, 64(1), 55-66, DOI: 10.1042/ebc20190053.
- Yang, J., Carroll, K.S., and Liebler, D.C., The Expanding Landscape of the Thiol Redox Proteome. Mol Cell Proteomics, 2016, 15(1), 1-11, DOI: 10.1074/mcp.O115.056051.
- Finkel, T., Signal transduction by reactive oxygen species. J Cell Biol, 2011, 194(1), 7-15, DOI: 10.1083/jcb.201102095.
- Szyller, J. and Bil-Lula, I., Heat Shock Proteins in Oxidative Stress and Ischemia/Reperfusion Injury and Benefits from Physical Exercises: A Review to the Current Knowledge. Oxid Med Cell Longev, 2021, 2021, 6678457, DOI: 10.1155/2021/6678457.
- Kalmar, B. and Greensmith, L., Induction of heat shock proteins for protection against oxidative stress. Adv Drug Deliv Rev, 2009, 61(4), 310-8, DOI: 10.1016/j.addr.2009.02.003.
- Yamamoto, M., Kensler, T.W., and Motohashi, H., The KEAP1-NRF2 System: a Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev, 2018, 98(3), 1169-1203, DOI: 10.1152/physrev.00023.2017.
- Korovila, I., Hugo, M., Castro, J.P., Weber, D., Höhn, A., Grune, T., and Jung, T., Proteostasis, oxidative stress and aging. Redox Biol, 2017, 13, 550-567, DOI: 10.1016/j.redox.2017.07.008.
- Fernández-Fernández, M.R. and Valpuesta, J.M., Hsp70 chaperone: a master player in protein homeostasis. F1000Res, 2018, 7, DOI: 10.12688/f1000research.15528.1.
- Large, A.T., Goldberg, M.D., and Lund, P.A., Chaperones and protein folding in the archaea. Biochem Soc Trans, 2009, 37(Pt 1), 46-51, DOI: 10.1042/bst0370046.
- Rebeaud, M.E., Mallik, S., Goloubinoff, P., and Tawfik, D.S., On the evolution of chaperones and cochaperones and the expansion of proteomes across the Tree of Life. Proc Natl Acad Sci U S A, 2021, 118(21), e2020885118 DOI: 10.1073/pnas.2020885118.
- Rosenzweig, R., Nillegoda, N.B., Mayer, M.P., and Bukau, B., The Hsp70 chaperone network. Nat Rev Mol Cell Biol, 2019, 20(11), 665-680, DOI: 10.1038/s41580-019-0133-3.
- Murphy, M.E., The HSP70 family and cancer. Carcinogenesis, 2013, 34(6), 1181-8, DOI: 10.1093/carcin/bgt111.
- Shrestha, L. and Young, J.C., Function and Chemotypes of Human Hsp70 Chaperones. Curr Top Med Chem, 2016, 16(25), 2812-28, DOI: 10.2174/1568026616666160413142028.
- Kabani, M. and Martineau, C.N., Multiple hsp70 isoforms in the eukaryotic cytosol: mere redundancy or functional specificity? Curr Genomics, 2008, 9(5), 338-248, DOI: 10.2174/138920208785133280.
- Radons, J., The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperones, 2016, 21(3), 379-404, DOI: 10.1007/s12192-016-0676-6.
- Bertelsen, E.B., Chang, L., Gestwicki, J.E., and Zuiderweg, E.R., Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A, 2009, 106(21), 8471-6, DOI: 10.1073/pnas.0903503106.
- Zhuravleva, A. and Gierasch, L.M., Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci U S A, 2011, 108(17), 6987-92, DOI: 10.1073/pnas.1014448108.
- Zhang, Y. and Zuiderweg, E.R., The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc Natl Acad Sci U S A, 2004, 101(28), 10272-7, DOI: 10.1073/pnas.0401313101.
- Zhang, P., Leu, J.I., Murphy, M.E., George, D.L., and Marmorstein, R., Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate. PLoS One, 2014, 9(7), e103518, DOI: 10.1371/journal.pone.0103518.
- Zhuravleva, A. and Gierasch, L.M., Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc Natl Acad Sci U S A, 2015, 112(22), E2865-73, DOI: 10.1073/pnas.1506692112.
- Xu, L., Gong, W., Cusack, S.A., Wu, H., Loovers, H.M., Zhang, H., Perrett, S., and Jones, G.W., The β6/β7 region of the Hsp70 substrate-binding domain mediates heat-shock response and prion propagation. Cell Mol Life Sci, 2018, 75(8), 1445-1459, DOI: 10.1007/s00018-017-2698-3.
- Xu, L., Zhang, H., Cuskelly, D.D., Doyle, S., Perrett, S., and Jones, G.W., Mutational analysis of the Hsp70 substrate-binding domain: Correlating molecular-level changes with in vivo function. Mol Microbiol, 2021, 115(6), 1262-1276, DOI: 10.1111/mmi.14671.
- Jiang, Y., Rossi, P., and Kalodimos, C.G., Structural basis for client recognition and activity of Hsp40 chaperones. Science, 2019, 365(6459), 1313-1319, DOI: 10.1126/science.aax1280.
- Matsumura, Y., Sakai, J., and Skach, W.R., Endoplasmic reticulum protein quality control is determined by cooperative interactions between Hsp/c70 protein and the CHIP E3 ligase. J Biol Chem, 2013, 288(43), 31069-79, DOI: 10.1074/jbc.M113.479345.
- Smock, R.G., Blackburn, M.E., and Gierasch, L.M., Conserved, disordered C terminus of DnaK enhances cellular survival upon stress and DnaK in vitro chaperone activity. J Biol Chem, 2011, 286(36), 31821-9, DOI: 10.1074/jbc.M111.265835.
- Gong, W., Hu, W., Xu, L., Wu, H., Wu, S., Zhang, H., Wang, J., Jones, G.W., and Perrett, S., The C-terminal GGAP motif of Hsp70 mediates substrate recognition and stress response in yeast. J Biol Chem, 2018, 293(46), 17663-17675, DOI: 10.1074/jbc.RA118.002691.
- Zuiderweg, E.R., Bertelsen, E.B., Rousaki, A., Mayer, M.P., Gestwicki, J.E., and Ahmad, A., Allostery in the Hsp70 chaperone proteins. Top Curr Chem, 2013, 328, 99-153, DOI: 10.1007/128_2012_323.
- Wu, S., Hong, L., Wang, Y., Yu, J., Yang, J., Yang, J., Zhang, H., and Perrett, S., Kinetics of the conformational cycle of Hsp70 reveals the importance of the dynamic and heterogeneous nature of Hsp70 for its function. Proc Natl Acad Sci U S A, 2020, 117(14), 7814-7823, DOI: 10.1073/pnas.1914376117.
- Kityk, R., Kopp, J., Sinning, I., and Mayer, M.P., Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell, 2012, 48(6), 863-74, DOI: 10.1016/j.molcel.2012.09.023.
- Qi, R., Sarbeng, E.B., Liu, Q., Le, K.Q., Xu, X., Xu, H., Yang, J., Wong, J.L., Vorvis, C., Hendrickson, W.A., Zhou, L., and Liu, Q., Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat Struct Mol Biol, 2013, 20(7), 900-7, DOI: 10.1038/nsmb.2583.
- Wang, W., Liu, Q., Liu, Q., and Hendrickson, W.A., Conformational equilibria in allosteric control of Hsp70 chaperones. Mol Cell, 2021, DOI: 10.1016/j.molcel.2021.07.039.
- Meng, W., Clerico, E.M., McArthur, N., and Gierasch, L.M., Allosteric landscapes of eukaryotic cytoplasmic Hsp70s are shaped by evolutionary tuning of key interfaces. Proc Natl Acad Sci U S A, 2018, 115(47), 11970-11975, DOI: 10.1073/pnas.1811105115.
- Zhuravleva, A., Clerico, E.M., and Gierasch, L.M., An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell, 2012, 151(6), 1296-307, DOI: 10.1016/j.cell.2012.11.002.
- Faust, O., Abayev-Avraham, M., Wentink, A.S., Maurer, M., Nillegoda, N.B., London, N., Bukau, B., and Rosenzweig, R., HSP40 proteins use class-specific regulation to drive HSP70 functional diversity. Nature, 2020, 587(7834), 489-494, DOI: 10.1038/s41586-020-2906-4.
- Polier, S., Dragovic, Z., Hartl, F.U., and Bracher, A., Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell, 2008, 133(6), 1068-79, DOI: 10.1016/j.cell.2008.05.022.
- Bracher, A. and Verghese, J., The nucleotide exchange factors of Hsp70 molecular chaperones. Front Mol Biosci, 2015, 2, 10, DOI: 10.3389/fmolb.2015.00010.
- Qiu, X.B., Shao, Y.M., Miao, S., and Wang, L., The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci, 2006, 63(22), 2560-70, DOI: 10.1007/s00018-006-6192-6.
- Kravats, A.N., Hoskins, J.R., Reidy, M., Johnson, J.L., Doyle, S.M., Genest, O., Masison, D.C., and Wickner, S., Functional and physical interaction between yeast Hsp90 and Hsp70. Proc Natl Acad Sci U S A, 2018, 115(10), E2210-e2219, DOI: 10.1073/pnas.1719969115.
- Winkler, J., Tyedmers, J., Bukau, B., and Mogk, A., Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol, 2012, 198(3), 387-404, DOI: 10.1083/jcb.201201074.
- Stankiewicz, M., Nikolay, R., Rybin, V., and Mayer, M.P., CHIP participates in protein triage decisions by preferentially ubiquitinating Hsp70-bound substrates. Febs j, 2010, 277(16), 3353-67, DOI: 10.1111/j.1742-4658.2010.07737.x.
- Dice, J.F., Chaperone-mediated autophagy. Autophagy, 2007, 3(4), 295-9, DOI: 10.4161/auto.4144.
- Yang, J., Zhang, H., Gong, W., Liu, Z., Wu, H., Hu, W., Chen, X., Wang, L., Wu, S., Chen, C., and Perrett, S., S-Glutathionylation of human inducible Hsp70 reveals a regulatory mechanism involving the C-terminal α-helical lid. J Biol Chem, 2020, 295(24), 8302-8324, DOI: 10.1074/jbc.RA119.012372.
- Grunwald, M.S., Pires, A.S., Zanotto-Filho, A., Gasparotto, J., Gelain, D.P., Demartini, D.R., Schöler, C.M., de Bittencourt, P.I., Jr., and Moreira, J.C., The oxidation of HSP70 is associated with functional impairment and lack of stimulatory capacity. Cell Stress Chaperones, 2014, 19(6), 913-25, DOI: 10.1007/s12192-014-0516-5.
- Wang, Y., Gibney, P.A., West, J.D., and Morano, K.A., The yeast Hsp70 Ssa1 is a sensor for activation of the heat shock response by thiol-reactive compounds. Mol Biol Cell, 2012, 23(17), 3290-8, DOI: 10.1091/mbc.E12-06-0447.
- Weerapana, E., Wang, C., Simon, G.M., Richter, F., Khare, S., Dillon, M.B., Bachovchin, D.A., Mowen, K., Baker, D., and Cravatt, B.F., Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature, 2010, 468(7325), 790-5, DOI: 10.1038/nature09472.
- Sun, M.A., Wang, Y., Cheng, H., Zhang, Q., Ge, W., and Guo, D., RedoxDB--a curated database for experimentally verified protein oxidative modification. Bioinformatics, 2012, 28(19), 2551-2, DOI: 10.1093/bioinformatics/bts468.
- Zhang, X., Huang, B., Zhang, L., Zhang, Y., Zhao, Y., Guo, X., Qiao, X., and Chen, C., SNObase, a database for S-nitrosation modification. Protein Cell, 2012, 3(12), 929-33, DOI: 10.1007/s13238-012-2094-6.
- Chen, Y.J., Lu, C.T., Lee, T.Y., and Chen, Y.J., dbGSH: a database of S-glutathionylation. Bioinformatics, 2014, 30(16), 2386-8, DOI: 10.1093/bioinformatics/btu301.
- Chen, Y.J., Lu, C.T., Su, M.G., Huang, K.Y., Ching, W.C., Yang, H.H., Liao, Y.C., Chen, Y.J., and Lee, T.Y., dbSNO 2.0: a resource for exploring structural environment, functional and disease association and regulatory network of protein S-nitrosylation. Nucleic Acids Res, 2015, 43(Database issue), D503-11, DOI: 10.1093/nar/gku1176.
- Wang, P., Zhang, Q., Li, S., Cheng, B., Xue, H., Wei, Z., Shao, T., Liu, Z.X., Cheng, H., and Wang, Z., iCysMod: an integrative database for protein cysteine modifications in eukaryotes. Brief Bioinform, 2021, 22(5), DOI: 10.1093/bib/bbaa400.
- Fratelli, M., Gianazza, E., and Ghezzi, P., Redox proteomics: identification and functional role of glutathionylated proteins. Expert Rev Proteomics, 2004, 1(3), 365-76, DOI: 10.1586/14789450.1.3.365.
- Zhang, H., Yang, J., Wu, S., Gong, W., Chen, C., and Perrett, S., Glutathionylation of the Bacterial Hsp70 Chaperone DnaK Provides a Link between Oxidative Stress and the Heat Shock Response. J Biol Chem, 2016, 291(13), 6967-81, DOI: 10.1074/jbc.M115.673608.
- Gamer, J., Multhaup, G., Tomoyasu, T., McCarty, J.S., Rüdiger, S., Schönfeld, H.J., Schirra, C., Bujard, H., and Bukau, B., A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. Embo j, 1996, 15(3), 607-17.
- Müller, A., Hoffmann, J.H., Meyer, H.E., Narberhaus, F., Jakob, U., and Leichert, L.I., Nonnative disulfide bond formation activates the σ32-dependent heat shock response in Escherichia coli. J Bacteriol, 2013, 195(12), 2807-16, DOI: 10.1128/jb.00127-13.
- Winter, J., Linke, K., Jatzek, A., and Jakob, U., Severe oxidative stress causes inactivation of DnaK and activation of the redox-regulated chaperone Hsp33. Mol Cell, 2005, 17(3), 381-92, DOI: 10.1016/j.molcel.2004.12.027.
- Chung, H.S., Murray, C.I., Venkatraman, V., Crowgey, E.L., Rainer, P.P., Cole, R.N., Bomgarden, R.D., Rogers, J.C., Balkan, W., Hare, J.M., Kass, D.A., and Van Eyk, J.E., Dual Labeling Biotin Switch Assay to Reduce Bias Derived From Different Cysteine Subpopulations: A Method to Maximize S-Nitrosylation Detection. Circ Res, 2015, 117(10), 846-57, DOI: 10.1161/circresaha.115.307336.
- Mnatsakanyan, R., Markoutsa, S., Walbrunn, K., Roos, A., Verhelst, S.H.L., and Zahedi, R.P., Proteome-wide detection of S-nitrosylation targets and motifs using bioorthogonal cleavable-linker-based enrichment and switch technique. Nat Commun, 2019, 10(1), 2195, DOI: 10.1038/s41467-019-10182-4.
- Lefievre, L., Chen, Y., Conner, S.J., Scott, J.L., Publicover, S.J., Ford, W.C., and Barratt, C.L., Human spermatozoa contain multiple targets for protein S-nitrosylation: an alternative mechanism of the modulation of sperm function by nitric oxide? Proteomics, 2007, 7(17), 3066-84, DOI: 10.1002/pmic.200700254.
- Huang, B., Chen, S.C., and Wang, D.L., Shear flow increases S-nitrosylation of proteins in endothelial cells. Cardiovasc Res, 2009, 83(3), 536-46, DOI: 10.1093/cvr/cvp154.
- Fu, L., Liu, K., He, J., Tian, C., Yu, X., and Yang, J., Direct Proteomic Mapping of Cysteine Persulfidation. Antioxid Redox Signal, 2020, 33(15), 1061-1076, DOI: 10.1089/ars.2019.7777.
- Wu, Q., Zhao, B., Weng, Y., Shan, Y., Li, X., Hu, Y., Liang, Z., Yuan, H., Zhang, L., and Zhang, Y., Site-Specific Quantification of Persulfidome by Combining an Isotope-Coded Affinity Tag with Strong Cation-Exchange-Based Fractionation. Anal Chem, 2019, 91(23), 14860-14864, DOI: 10.1021/acs.analchem.9b04112.
- Miyata, Y., Rauch, J.N., Jinwal, U.K., Thompson, A.D., Srinivasan, S., Dickey, C.A., and Gestwicki, J.E., Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem Biol, 2012, 19(11), 1391-9, DOI: 10.1016/j.chembiol.2012.07.026.
- Fu, L., Liu, K., Ferreira, R.B., Carroll, K.S., and Yang, J., Proteome-Wide Analysis of Cysteine S-Sulfenylation Using a Benzothiazine-Based Probe. Curr Protoc Protein Sci, 2019, 95(1), e76, DOI: 10.1002/cpps.76.
- Yang, J., Gupta, V., Carroll, K.S., and Liebler, D.C., Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat Commun, 2014, 5, 4776, DOI: 10.1038/ncomms5776.
- Akter, S., Fu, L., Jung, Y., Conte, M.L., Lawson, J.R., Lowther, W.T., Sun, R., Liu, K., Yang, J., and Carroll, K.S., Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat Chem Biol, 2018, 14(11), 995-1004, DOI: 10.1038/s41589-018-0116-2.
- Valek, L., Heidler, J., Scheving, R., Wittig, I., and Tegeder, I., Nitric oxide contributes to protein homeostasis by S-nitrosylations of the chaperone HSPA8 and the ubiquitin ligase UBE2D. Redox Biol, 2019, 20, 217-235, DOI: 10.1016/j.redox.2018.10.002.
- Wang, J. and Sevier, C.S., Formation and Reversibility of BiP Protein Cysteine Oxidation Facilitate Cell Survival during and post Oxidative Stress. J Biol Chem, 2016, 291(14), 7541-57, DOI: 10.1074/jbc.M115.694810.
- Flohé, L., The fairytale of the GSSG/GSH redox potential. Biochim Biophys Acta, 2013, 1830(5), 3139-42, DOI: 10.1016/j.bbagen.2012.10.020.
- Wang, J., Pareja, K.A., Kaiser, C.A., and Sevier, C.S., Redox signaling via the molecular chaperone BiP protects cells against endoplasmic reticulum-derived oxidative stress. Elife, 2014, 3, e03496, DOI: 10.7554/eLife.03496.
- Xu, M., Marsh, H.M., and Sevier, C.S., A Conserved Cysteine within the ATPase Domain of the Endoplasmic Reticulum Chaperone BiP is Necessary for a Complete Complement of BiP Activities. J Mol Biol, 2016, 428(20), 4168-4184, DOI: 10.1016/j.jmb.2016.08.011.
- Breitzig, M., Bhimineni, C., Lockey, R., and Kolliputi, N., 4-Hydroxy-2-nonenal: a critical target in oxidative stress? Am J Physiol Cell Physiol, 2016, 311(4), C537-c543, DOI: 10.1152/ajpcell.00101.2016.
- Chaudhary, P., Sharma, R., Sharma, A., Vatsyayan, R., Yadav, S., Singhal, S.S., Rauniyar, N., Prokai, L., Awasthi, S., and Awasthi, Y.C., Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry, 2010, 49(29), 6263-75, DOI: 10.1021/bi100517x.
- Jaganjac, M., Cindrić, M., Jakovčević, A., Žarković, K., and Žarković, N., Lipid peroxidation in brain tumors. Neurochem Int, 2021, 149, 105118, DOI: 10.1016/j.neuint.2021.105118.
- Reyes-Jiménez, E., Ramírez-Hernández, A.A., Santos-Álvarez, J.C., Velázquez-Enríquez, J.M., Pina-Canseco, S., Baltiérrez-Hoyos, R., and Vásquez-Garzón, V.R., Involvement of 4-hydroxy-2-nonenal in the pathogenesis of pulmonary fibrosis. Mol Cell Biochem, 2021, DOI: 10.1007/s11010-021-04244-9.
- Skorokhod, O.A., Davalos-Schafler, D., Gallo, V., Valente, E., Ulliers, D., Notarpietro, A., Mandili, G., Novelli, F., Persico, M., Taglialatela-Scafati, O., Arese, P., and Schwarzer, E., Oxidative stress-mediated antimalarial activity of plakortin, a natural endoperoxide from the tropical sponge Plakortis simplex. Free Radic Biol Med, 2015, 89, 624-37, DOI: 10.1016/j.freeradbiomed.2015.10.399.
- Carbone, D.L., Doorn, J.A., Kiebler, Z., Sampey, B.P., and Petersen, D.R., Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem Res Toxicol, 2004, 17(11), 1459-67, DOI: 10.1021/tx049838g.
- Galligan, J.J., Fritz, K.S., Backos, D.S., Shearn, C.T., Smathers, R.L., Jiang, H., MacLean, K.N., Reigan, P.R., and Petersen, D.R., Oxidative stress-mediated aldehyde adduction of GRP78 in a mouse model of alcoholic liver disease: functional independence of ATPase activity and chaperone function. Free Radic Biol Med, 2014, 73, 411-20, DOI: 10.1016/j.freeradbiomed.2014.06.002.
- Yang, L.L., Chen, H., Wang, J., Xia, T., Sun, H., Yuan, C.H., Liu, S.L., and Chen, J.B., 4-HNE Induces Apoptosis of Human Retinal Pigment Epithelial Cells by Modifying HSP70. Curr Med Sci, 2019, 39(3), 442-448, DOI: 10.1007/s11596-019-2057-8.
- Polla, B.S., Kantengwa, S., François, D., Salvioli, S., Franceschi, C., Marsac, C., and Cossarizza, A., Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proc Natl Acad Sci U S A, 1996, 93(13), 6458-63, DOI: 10.1073/pnas.93.13.6458.
- Khomenko, I.P., Bakhtina, L.Y., Zelenina, O.M., Kruglov, S.V., Manukhina, E.B., Bayda, L.A., and Malyshev, I.Y., Role of heat shock proteins HSP70 and HSP32 in the protective effect of adaptation of cultured HT22 hippocampal cells to oxidative stress. Bull Exp Biol Med, 2007, 144(2), 174-7, DOI: 10.1007/s10517-007-0282-9.
- Watanabe, S., Ageta-Ishihara, N., Nagatsu, S., Takao, K., Komine, O., Endo, F., Miyakawa, T., Misawa, H., Takahashi, R., Kinoshita, M., and Yamanaka, K., SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol Brain, 2014, 7, 62, DOI: 10.1186/s13041-014-0062-1.
- Guo, S., Wharton, W., Moseley, P., and Shi, H., Heat shock protein 70 regulates cellular redox status by modulating glutathione-related enzyme activities. Cell Stress Chaperones, 2007, 12(3), 245-54, DOI: 10.1379/csc-265.1.
- Broome, C.S., Kayani, A.C., Palomero, J., Dillmann, W.H., Mestril, R., Jackson, M.J., and McArdle, A., Effect of lifelong overexpression of HSP70 in skeletal muscle on age-related oxidative stress and adaptation after nondamaging contractile activity. Faseb j, 2006, 20(9), 1549-51, DOI: 10.1096/fj.05-4935fje.
- Xu, L., Voloboueva, L.A., Ouyang, Y., Emery, J.F., and Giffard, R.G., Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab, 2009, 29(2), 365-74, DOI: 10.1038/jcbfm.2008.125.
- Chen, B.H., Chang, Y.J., Lin, S., and Yang, W.Y., Hsc70/Stub1 promotes the removal of individual oxidatively stressed peroxisomes. Nat Commun, 2020, 11(1), 5267, DOI: 10.1038/s41467-020-18942-3.
- Li, X., Yu, Y., Gorshkov, B., Haigh, S., Bordan, Z., Weintraub, D., Rudic, R.D., Chakraborty, T., Barman, S.A., Verin, A.D., Su, Y., Lucas, R., Stepp, D.W., Chen, F., and Fulton, D.J.R., Hsp70 Suppresses Mitochondrial Reactive Oxygen Species and Preserves Pulmonary Microvascular Barrier Integrity Following Exposure to Bacterial Toxins. Front Immunol, 2018, 9, 1309, DOI: 10.3389/fimmu.2018.01309.
- Pistono, C., Monti, M.C., Boiocchi, C., Berzolari, F.G., Osera, C., Mallucci, G., Cuccia, M., Pascale, A., Montomoli, C., and Bergamaschi, R., Response to oxidative stress of peripheral blood mononuclear cells from multiple sclerosis patients and healthy controls. Cell Stress Chaperones, 2020, 25(1), 81-91, DOI: 10.1007/s12192-019-01049-0.
- Yan, L.J., Christians, E.S., Liu, L., Xiao, X., Sohal, R.S., and Benjamin, I.J., Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. Embo j, 2002, 21(19), 5164-72, DOI: 10.1093/emboj/cdf528.
- Dayalan Naidu, S., Kostov, R.V., and Dinkova-Kostova, A.T., Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol Sci, 2015, 36(1), 6-14, DOI: 10.1016/j.tips.2014.10.011.
- Girard, P.M., Peynot, N., and Lelièvre, J.M., Differential correlations between changes to glutathione redox state, protein ubiquitination, and stress-inducible HSPA chaperone expression after different types of oxidative stress. Cell Stress Chaperones, 2018, 23(5), 985-1002, DOI: 10.1007/s12192-018-0909-y.
- Doulias, P.T., Kotoglou, P., Tenopoulou, M., Keramisanou, D., Tzavaras, T., Brunk, U., Galaris, D., and Angelidis, C., Involvement of heat shock protein-70 in the mechanism of hydrogen peroxide-induced DNA damage: the role of lysosomes and iron. Free Radic Biol Med, 2007, 42(4), 567-77, DOI: 10.1016/j.freeradbiomed.2006.11.022.
- Adachi, M., Liu, Y., Fujii, K., Calderwood, S.K., Nakai, A., Imai, K., and Shinomura, Y., Oxidative stress impairs the heat stress response and delays unfolded protein recovery. PLoS One, 2009, 4(11), e7719, DOI: 10.1371/journal.pone.0007719.
- Bianchi, A., Moulin, D., Hupont, S., Koufany, M., Netter, P., Reboul, P., and Jouzeau, J.Y., Oxidative stress-induced expression of HSP70 contributes to the inhibitory effect of 15d-PGJ2 on inducible prostaglandin pathway in chondrocytes. Free Radic Biol Med, 2014, 76, 114-26, DOI: 10.1016/j.freeradbiomed.2014.07.028.
- Reeg, S., Jung, T., Castro, J.P., Davies, K.J.A., Henze, A., and Grune, T., The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic Biol Med, 2016, 99, 153-166, DOI: 10.1016/j.freeradbiomed.2016.08.002.
- Kiffin, R., Christian, C., Knecht, E., and Cuervo, A.M., Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell, 2004, 15(11), 4829-40, DOI: 10.1091/mbc.e04-06-0477.
- Dodson, M., Darley-Usmar, V., and Zhang, J., Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med, 2013, 63, 207-21, DOI: 10.1016/j.freeradbiomed.2013.05.014.
- Zemanovic, S., Ivanov, M.V., Ivanova, L.V., Bhatnagar, A., Michalkiewicz, T., Teng, R.J., Kumar, S., Rathore, R., Pritchard, K.A., Jr., Konduri, G.G., and Afolayan, A.J., Dynamic Phosphorylation of the C Terminus of Hsp70 Regulates the Mitochondrial Import of SOD2 and Redox Balance. Cell Rep, 2018, 25(9), 2605-2616.e7, DOI: 10.1016/j.celrep.2018.11.015.
- Xu, N.W., Chen, Y., Liu, W., Chen, Y.J., Fan, Z.M., Liu, M., and Li, L.J., Inhibition of JAK2/STAT3 Signaling Pathway Suppresses Proliferation of Burkitt's Lymphoma Raji Cells via Cell Cycle Progression, Apoptosis, and Oxidative Stress by Modulating HSP70. Med Sci Monit, 2018, 24, 6255-6263, DOI: 10.12659/msm.910170.
- Hensen, S.M., Heldens, L., van Enckevort, C.M., van Genesen, S.T., Pruijn, G.J., and Lubsen, N.H., Activation of the antioxidant response in methionine deprived human cells results in an HSF1-independent increase in HSPA1A mRNA levels. Biochimie, 2013, 95(6), 1245-51, DOI: 10.1016/j.biochi.2013.01.017.
- Smith, M.R., Fernandes, J., Go, Y.M., and Jones, D.P., Redox dynamics of manganese as a mitochondrial life-death switch. Biochem Biophys Res Commun, 2017, 482(3), 388-398, DOI: 10.1016/j.bbrc.2016.10.126.
- Afolayan, A.J., Teng, R.J., Eis, A., Rana, U., Broniowska, K.A., Corbett, J.A., Pritchard, K., and Konduri, G.G., Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am J Physiol Lung Cell Mol Physiol, 2014, 306(4), L351-60, DOI: 10.1152/ajplung.00264.2013.
- Zhang, H., Amick, J., Chakravarti, R., Santarriaga, S., Schlanger, S., McGlone, C., Dare, M., Nix, J.C., Scaglione, K.M., Stuehr, D.J., Misra, S., and Page, R.C., A bipartite interaction between Hsp70 and CHIP regulates ubiquitination of chaperoned client proteins. Structure, 2015, 23(3), 472-482, DOI: 10.1016/j.str.2015.01.003.
- Xie, Y., Shi, X., Sheng, K., Han, G., Li, W., Zhao, Q., Jiang, B., Feng, J., Li, J., and Gu, Y., PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol Med Rep, 2019, 19(2), 783-791, DOI: 10.3892/mmr.2018.9713.
- Lu, R., Zhang, Y.G., and Sun, J., STAT3 activation in infection and infection-associated cancer. Mol Cell Endocrinol, 2017, 451, 80-87, DOI: 10.1016/j.mce.2017.02.023.
- Alas, S. and Bonavida, B., Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin's lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin Cancer Res, 2003, 9(1), 316-26.
- Madamanchi, N.R., Li, S., Patterson, C., and Runge, M.S., Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler Thromb Vasc Biol, 2001, 21(3), 321-6, DOI: 10.1161/01.atv.21.3.321.
- Marinho, H.S., Real, C., Cyrne, L., Soares, H., and Antunes, F., Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol, 2014, 2, 535-62, DOI: 10.1016/j.redox.2014.02.006.
- Ali, A., Bharadwaj, S., O'Carroll, R., and Ovsenek, N., HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol, 1998, 18(9), 4949-60, DOI: 10.1128/mcb.18.9.4949.
- Shi, Y., Mosser, D.D., and Morimoto, R.I., Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev, 1998, 12(5), 654-66, DOI: 10.1101/gad.12.5.654.
- Kmiecik, S.W. and Mayer, M.P., Molecular mechanisms of heat shock factor 1 regulation. Trends Biochem Sci, 2021, DOI: 10.1016/j.tibs.2021.10.004.
- Paul, S., Ghosh, S., Mandal, S., Sau, S., and Pal, M., NRF2 transcriptionally activates the heat shock factor 1 promoter under oxidative stress and affects survival and migration potential of MCF7 cells. J Biol Chem, 2018, 293(50), 19303-19316, DOI: 10.1074/jbc.RA118.003376.
- Zhang, Y., Ahn, Y.H., Benjamin, I.J., Honda, T., Hicks, R.J., Calabrese, V., Cole, P.A., and Dinkova-Kostova, A.T., HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem Biol, 2011, 18(11), 1355-61, DOI: 10.1016/j.chembiol.2011.09.008.
- Hensen, S.M., Heldens, L., van Genesen, S.T., Pruijn, G.J., and Lubsen, N.H., A delayed antioxidant response in heat-stressed cells expressing a non-DNA binding HSF1 mutant. Cell Stress Chaperones, 2013, 18(4), 455-73, DOI: 10.1007/s12192-012-0400-0.