Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Vicky Zhou and Version 2 by Vicky Zhou.
Alcohol consumption is linked to the risk of prostate cancer (PCa). High alcohol intake, especially binge drinking, is associated with increased risk for PCa, and this effect is not limited to any type of beverage. Alcohol consumption is also directly linked to PCa lethality as it may accelerate the growth of prostate tumors and significantly shorten the time for the progression to metastatic PCa.
- alcohol consumption
- prostate cancer
- prostate cancer-associated mortality
1. Introduction
The link between alcohol consumption and malignant diseases has long intrigued researchers. In a review of 140 references from 1966 to 2020, mainly epidemiological studies, moderate alcohol consumption was correlated with increased risk of upper digestive tract, liver, colorectal, breast, pancreatic, and prostate cancers (PCa) [1]. Despite generally increased cancer risk from any type of alcohol, many patients continue to drink alcohol [2], and some governments continue to leave their citizens uninformed of the carcinogenic risk of alcohol consumption [3]. In a recent report, the World Health Organization (WHO) estimated that alcohol use resulted in 3 million deaths globally in 2016, 12.6% of those were associated with malignant neoplasms [4]. Among men, alcohol intake is the fourth-largest contributor to cancer [5].
In 2019, 25.8% of Americans ages 18 or older reported that they engaged in binge drinking in the past month, and another 6.3% reported that they consumed alcohol at a heavy level in the past month [6]. According to the 2019 National Survey on Drug Use and Health (NSDUH), 14.1 million US adults ages 18 and older (5.6% of this age group) had Alcohol Use Disorder (AUD) [7]. This includes 8.9 million men (7.3% of men in this age group) and 5.2 million women (4.0% of women in this age group). Among PCa patients from the 2012–2017 National Health Interview Survey (NHIS), the distribution of alcohol consumption was as follows: 4.2%—heavy drinkers (>14 drinks per week), 47.1%—light/moderate drinkers (up to 14 drinks per week), 12.2%—infrequent drinkers (1–11 drinks in the past year), 25.7%—former drinkers (≥12 drinks in a lifetime but with 0 drinks in the past year), and 10.8%—never drinkers (<12 drinks in a lifetime) [8].
2. The Impact of Alcohol on the Aggressiveness of Prostate Tumors and PCa-Associated Death
Chronic alcohol consumption has been shown to induce high-grade PCa and metastasis [9][10][11][12][13][14]. One US-based study found that a higher frequency of alcohol intake was associated with increased mortality from PCa [15]. In another survey, Canadian researchers interviewed PCa patients immediately after diagnosis and 2–3 years later to monitor the level of alcohol consumption; mortality data were collected for up to 19 years [14]. They found that patients who consumed more than eight drinks weekly had higher mortality than patients who stopped drinking after being diagnosed with PCa.
Observations from Australian patients indicate that a beer intake frequency of ≥5 days per week was associated with an increased risk of advanced PCa (histological Gleason score eight or higher) [16]. Similarly, an extensive analysis among the Japanese population found a positive association of alcohol consumption with PCa in subjects with advanced disease: compared to non-drinkers, increased risks were observed for those who consumed 0–149 g/week, 150–299 g/week, and ≥300 g/week [17].
A recent study aimed to analyze the link between the total intake of alcohol and death from cancer. Using a large international dataset from the United Nations (UN), Food and Agriculture Organization of the United Nations (FAO), Organization for Economic Co-operation and Development (OECD), World Bank, WHO, U.S. Department of Agriculture, U.S. Department of Health, and Eurobarometer, authorserein came to the unequivocal conclusion that alcohol is “significantly and positively associated with prevalence and mortality from total, colon, lung, breast, and prostate cancers” [18].
Compared to men without early-life use of alcohol, military veterans with heavier alcohol consumption earlier in life were more likely to be diagnosed with high-grade PCa [19]. Recent US-based analysis also suggested that states with more restrictive alcohol policies had lower alcohol-attributable PCa mortality [20]. Accordingly, the authorsrein stated that “strengthening alcohol control policies may be a promising cancer prevention strategy”. These conclusions are reflected in another US-based study which stated that reducing alcohol intake below one drink per day is associated with a significant decrease in risk of PCa-related death [21]. Similarly, an investigation from Japan found PCa-related death among daily drinkers increased 2.5 times [22].
Significantly, PCa mortality among Mormons and Seventh-day Adventists from Germany, who usually consumed alcohol at a low level, is reduced considerably compared with PCa patients from the best German cancer registry (Saarland) [23]. Interestingly, countries with the lowest mortality rate for PCa (Bhutan, Nepal, Bangladesh, North Korea, Turkmenistan, Uzbekistan, Sri Lanka, Tajikistan, and Yemen [24]) are also characterized by the lowest levels of alcohol consumption [25]. Another recent study analyzed PCa mortality data obtained from 71 countries through the International Agency for Research on Cancer and the Food and Agricultural Organization of the United Nations. AuthoHersein found a correlation between increased cases of PCa-related deaths and total animal fat calories, meat, milk, sugar, alcoholic beverages, and stimulants [26].
A recent Mendelian randomization study from the international PRACTICAL Consortium found that SNP within the ALDH1B1 gene was associated with mortality in men with low-grade PCa [27]. The auIthors of this manuscript c is concluded: “Reducing alcohol consumption could slow prostate cancer disease progression.” Another study of 3306 PCa patients with European ancestry from the PCa Consortium aimed to examine alcohol intake and SNPs in the four pathways associated with PCa aggressiveness (angiogenesis, mitochondria, miRNA, and androgen metabolism-related pathways) [28]. Investigators found that excessive alcohol intake significantly impacts PCa aggressiveness within the following genetic subgroups: CAMK2D (calcium/calmodulin-dependent protein kinase II delta, enriched in the cell cycle and the calcium signaling pathway), PRKCA (protein kinase C alpha, the modulator of cell adhesion and transformation), and ROBO1 (roundabout guidance receptor 1, cell migration factor and a tumor suppressor gene). In addition, ithe author is observed an additional moderate link between alcohol and SNPs of genes associated with PCa aggressiveness. These are HGF (hepatocyte growth factor), PDGFB (platelet-derived growth factor subunit B), SYK (spleen-associated tyrosine kinase), PDGFD (platelet-derived growth factor D), and COL4A3 (collagen type IV alpha 3 chain) [28].
It has been shown that SNPs in the TNF-α and IL-10 genes were associated with an increased PCa risk [29][30]. A recent study from the Indian population (105 PCa cases along with 115 control) found that an increasing percentage of TNF-α and IL-10 haplotypes were found to be positively associated with aggressiveness of PCa and alcohol consumption [31].
3. The Cellular Mechanisms of Alcohol’s Effect on the Progression of PCa
In recent years, it has become clear that alcohol has multiple damaging effects on intracellular organization, transportation, and structure of organelles [32]. Both acute and chronic alcohol exposure results in disorganization of Golgi and inhibition of both intra-Golgi and Golgi-to-plasma membrane trafficking. This, in turn, leads to intracellular accumulation of newly synthesized proteins and subsequent endoplasmic reticulum (ER) stress [33][34][35][36][37][38][39]. In addition, Ethanol (EtOH) and its metabolites alter the activity of different Rab proteins, the conductors of post-Golgi transportation, and endocytosis [40][41][42][43][44][45][46][47][48]. OurThe laboratory revealed that alcohol-induced Golgi disorganization is associated with impairment of both coat protein I (COPI) and coat protein II (COPII) vesiculation and monomerization of the Golgi scaffold proteins, golgins [34][49]. Dimerization of giantin, the largest golgin, appears to be essential for the post-alcohol recovery of Golgi in hepatocytes [50]. Interestingly, giantin also loses its dimeric conformation in advanced PCa cells, which is associated with severe Golgi fragmentation and translocation of critical Golgi glycosyltransferases from the Golgi to the ER [51][52][53]. Recent investigations indicated that Golgi disorganization is a hallmark of cancer progression [54][55][56][57]. WThe group introduced the concept of an “onco-Golgi”, which postulates that in different types of cancer, including PCa, the activation of many pro-oncogenic and pro-metastatic pathways is caused by the mislocalization of resident Golgi enzymes due to disorganization of this organelle [58]. WThe rgroup recently reported that in androgen-responsive PCa cells exposed to EtOH, Golgi disorganization is triggered by acetaldehyde (ACH) and results in enhanced anchorage-independent growth, adherence, migration, and secretion of prostate specific antigen (PSA) [59]. Alteration of the Golgi morphology leads to the translocation of glycogen synthase kinase β (GSK3β) from the Golgi to the cytoplasm, followed by the phosphorylation of histone deacetylase 6 (HDAC6) and activation of the downstream heat shock protein 90 (HSP90)–androgen receptor (AR) pathway [59] (Figure 1A). Therefore, EtOH may accelerate AR transactivation, the driver of prostate carcinogenesis. Significantly, EtOH administration promoted the growth of LNCaP-derived xenograft tumors in mice.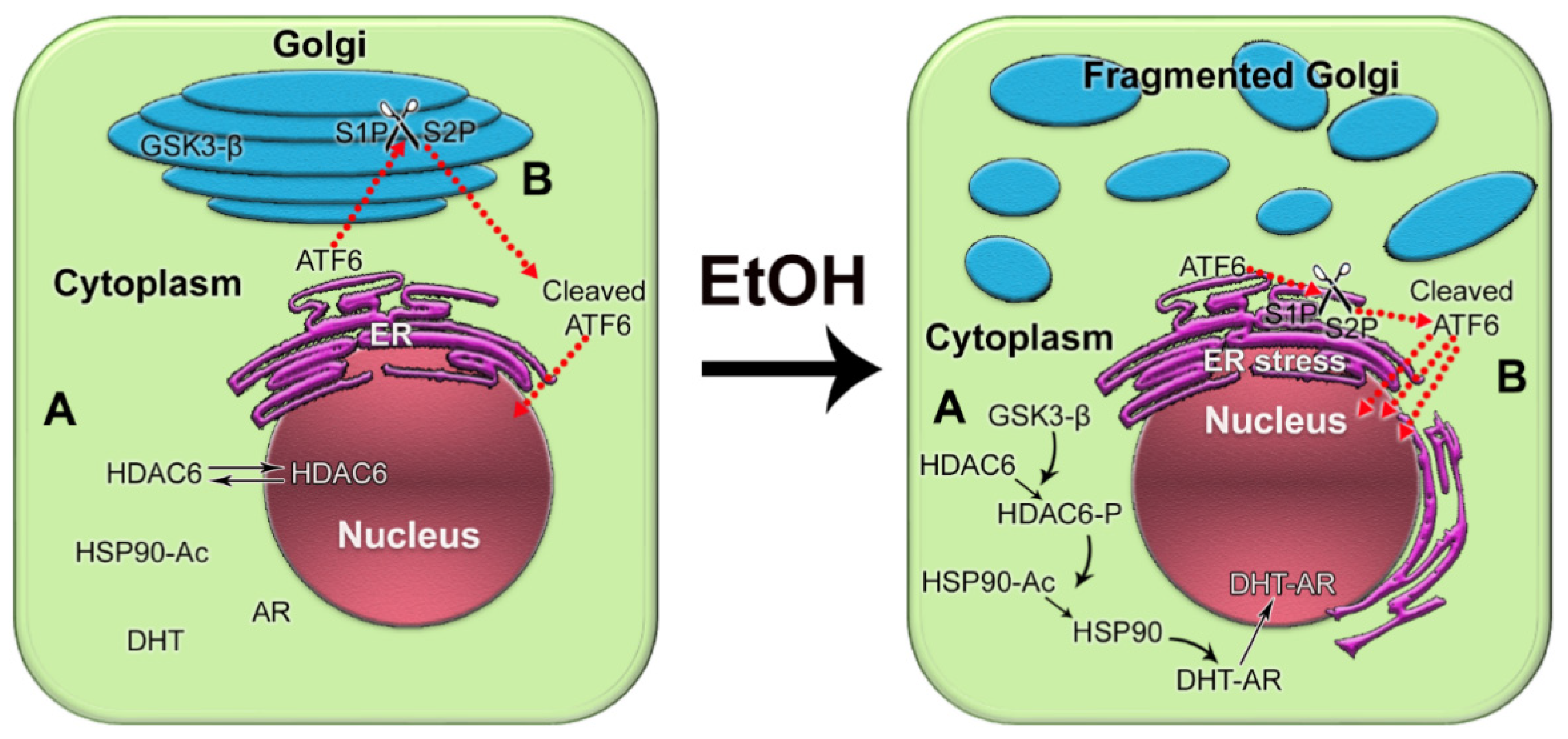
Figure 1. The impact of alcohol-induced Golgi disorganization on prostate carcinogenesis. (A) In normal prostate cells or low aggressive PCa cells, HDAC6 is distributed in both the nucleus and the cytoplasm. Typically, the phosphorylation of HDAC6 is moderate because the enzyme that phosphorylates HDAC6, GSK3β, is sequestered primarily within the Golgi. The acetylated HSP90 has a limited binding capacity to AR. EtOH treatment results in Golgi fragmentation and translocation of GSK3β to the cytoplasm, which results in increased phosphorylation of HDAC6. HDAC6-P deacetylates HSP90, which, in turn, accelerates conformational maturation of AR, its binding to dihydrotestosterone (DHT), and translocation to the nucleus. (B) In normal prostate and low-aggressive PCa cells, ATF6α is cleaved sequentially in the Golgi by S1P and S2P proteases. The dimeric form of trans-golgin GCC185 is the retention partner for both S1P and S2P. Cleaved ATF6 enters the nucleus and binds to ER stress-response elements, stimulating the expression of UPR genes. EtOH and its metabolites fragment Golgi membranes, which is associated with the monomerization of GCC185 and the subsequent shift of S1P and S2P to the ER. This simplifies and accelerates ATF6 cleavage, resulting in more prominent UPR signaling to maintain tumor cell growth and proliferation.
4. Conclusions
Despite the limitations discussed here, the link between alcohol consumption and the development of PCa is strong. Still, PCa development depends critically on other factors, notably diet, smoking, age, race (black men have higher incidence and mortality than white men), physical and sexual activity, stress, obesity, family history of PCa, and chronic prostatitis [76][77][78] (Figure 2A). For instance, the risk of PCa associated with the pro-inflammatory potential of the diet is accelerated in low-to-moderate alcohol drinkers [79]. In addition, alcohol intake was directly associated with PCa risk among individuals with lower dietary fiber intake and low folate intake [80][81].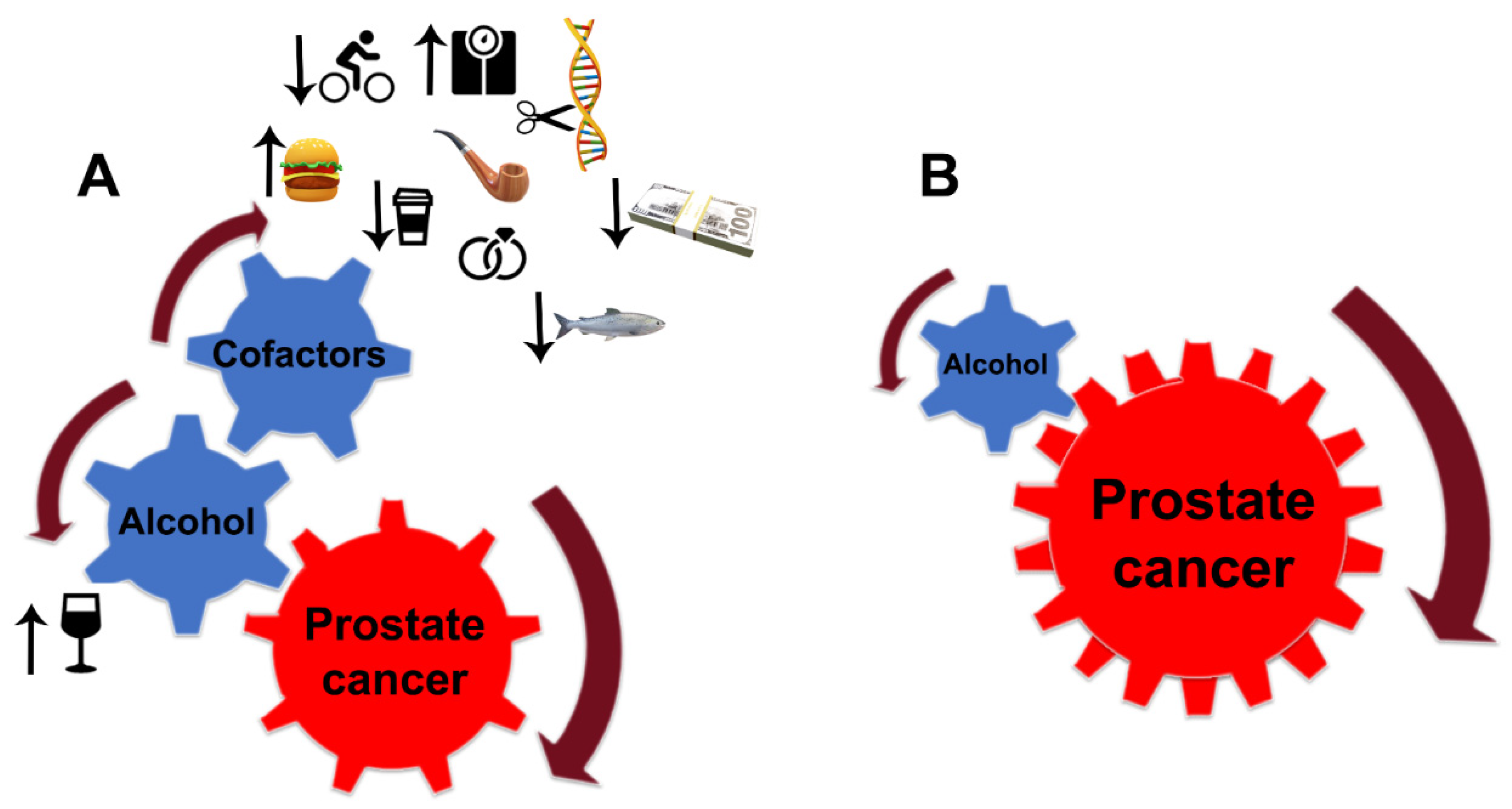
Figure 2. Alcohol interference in the development and progression of PCa. (A) Alcohol is a critical player and driver of prostate carcinogenesis. The carcinogenic effects of EtOH and its metabolites are magnified by multiple cofactors, such as obesity, smoking, excessive high-fat and red meat diet, low-level consumption of fish, caffeine, and linoleic acid, low physical activity, SNP of alcohol-related genes, and family history of PCa. Additional factors may include income and marital status: unmarried patients with an unstable financial situation are at higher risk of PCa. (B) In patients diagnosed with PCa, alcohol’s contribution to prostate tumor progression does not require cofactors. EtOH metabolites are sufficient to drive tumor growth and raise the metastatic potential of cancer cells.
References
- Teissedre, P.-L.; Rasines-Perea, Z.; Ruf, J.-C.; Stockley, C.; Antoce, A.O.; Romano, R.; Fradera, U.; Kosti, R.I. Effects of alcohol consumption in general, and wine in particular, on the risk of cancer development: A review. OENO One 2020, 54, 813–832.
- Agarwal, S.K. Alcohol and noncommunicable diseases: Part ii cancer, diabetes mellitus, kidney diseases, Alzheimer’s disease, arthritis. CMI 2021, 2, 185–200.
- Stockwell, T.; Solomon, R.; O’Brien, P.; Vallance, K.; Hobin, E. Cancer warning labels on alcohol containers: A consumer’s right to know, a government’s responsibility to inform, and an industry’s power to thwart. J. Stud. Alcohol. Drugs 2020, 81, 284–292.
- World Health Organization. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2018.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Substance Abuse and Mental Health Services Administration (Samhsa). Table 2.1b—Tobacco Product and Alcohol Use in Lifetime, Past Year, and Past Month among Persons Aged 12 or Older, by Age Group: Percentages, 2018 and 2019; National Survey on Drug Use and Health (NSDUH). 2019. Available online: https://www.samhsa.gov/data/sites/default/files/reports/rpt29394/NSDUHDetailedTabs2019/NSDUHDetTabsSect2pe2019.htm#tab2-1b (accessed on 12 January 2022).
- NIH. Understanding Alcohol Use Disorder. 2021. Available online: https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/understanding-alcohol-use-disorder (accessed on 12 January 2022).
- Lin, H.-Y.; Fisher, P.; Harris, D.; Tseng, T.-S. Alcohol Intake Patterns for Cancer and Non-Cancer Individuals: A population study. Transl. Cancer Res. 2019, 8, S334–S345.
- Gong, Z.; Kristal, A.R.; Schenk, J.M.; Tangen, C.M.; Goodman, P.J.; Thompson, I.M. Alcohol consumption, finasteride, and prostate cancer risk: Results from the prostate cancer prevention trial. Cancer 2009, 115, 3661–3669.
- Dickerman, B.A.; Markt, S.C.; Koskenvuo, M.; Pukkala, E.; Mucci, L.A.; Kaprio, J. Alcohol intake, drinking patterns, and prostate cancer risk and mortality: A 30-year prospective cohort study of Finnish twins. Cancer Causes Control 2016, 27, 1049–1058.
- Zuccolo, L.; Lewis, S.J.; Donovan, J.L.; Hamdy, F.C.; Neal, D.E.; Smith, G.D. Alcohol consumption and psa-detected prostate cancer risk—A case-control nested in the protect study. Int. J. Cancer 2013, 132, 2176–2185.
- Fowke, J.H.; Howard, L.; Andriole, G.L.; Freedland, S.J. Alcohol Intake Increases High-grade Prostate Cancer Risk Among Men Taking Dutasteride in the REDUCE Trial. Eur. Urol. 2014, 66, 1133–1138.
- Sawada, N.; Inoue, M.; Iwasaki, M.; Sasazuki, S.; Yamaji, T.; Shimazu, T.; Tsugane, S. Alcohol and smoking and subsequent risk of prostate cancer in Japanese men: The Japan Public Health Center-based prospective study. Int. J. Cancer 2013, 134, 971–978.
- Farris, M.S.; Courneya, K.S.; Kopciuk, K.A.; McGregor, S.E.; Friedenreich, C.M. Post-diagnosis alcohol intake and prostate cancer survival: A population-based cohort study. Int. J. Cancer 2018, 143, 253–262.
- Breslow, R.A.; Chen, C.M.; Graubard, B.I.; Mukamal, K.J. Prospective Study of Alcohol Consumption Quantity and Frequency and Cancer-Specific Mortality in the US Population. Am. J. Epidemiol. 2011, 174, 1044–1053.
- Papa, N.P.; MacInnis, R.J.; Jayasekara, H.; English, D.R.; Bolton, D.; Davis, I.D.; Lawrentschuk, N.; Millar, J.L.; Pedersen, J.; Severi, G.; et al. Total and beverage-specific alcohol intake and the risk of aggressive prostate cancer: A case–control study. Prostate Cancer Prostatic Dis. 2017, 20, 305–310.
- Sawada, N. Risk and preventive factors for prostate cancer in Japan: The Japan Public Health Center-based prospective (JPHC) study. J. Epidemiol. 2017, 27, 2–7.
- Penuelas, J.; Krisztin, T.; Obersteiner, M.; Huber, F.; Winner, H.; Janssens, I.A.; Ciais, P.; Sardans, J. Country-Level Relationships of the Human Intake of N and P, Animal and Vegetable Food, and Alcoholic Beverages with Cancer and Life Expectancy. Int. J. Environ. Res. Public Health 2020, 17, 7240.
- Michael, J.; Howard, L.E.; Markt, S.; De Hoedt, A.; Bailey, C.; Mucci, L.A.; Freedland, S.J.; Allott, E.H. Early-Life Alcohol Intake and High-Grade Prostate Cancer: Results from an Equal-Access, Racially Diverse Biopsy Cohort. Cancer Prev. Res. 2018, 11, 621–628.
- Alattas, M.; Ross, C.S.; Henehan, E.R.; Naimi, T.S. Alcohol policies and alcohol-attributable cancer mortality in U.S. States. Chem. Interact. 2019, 315, 108885.
- Kunzmann, A.T.; Coleman, H.G.; Huang, W.-Y.; Berndt, S.I. The association of lifetime alcohol use with mortality and cancer risk in older adults: A cohort study. PLoS Med. 2018, 15, e1002585.
- Hirayama, T. Life-style and cancer: From epidemiological evidence to public behavior change to mortality reduction of target cancers. J. Natl. Cancer Inst. Monogr. 1992, 12, 65–74.
- Grundmann, E. Cancer morbidity and mortality in USA Mormons and Seventh-day Adventists. Arch Anat. Cytol. Pathol. 1992, 40, 73–78.
- Hassanipour, S.; Mohammadian-Hafshejani, A.; Ghoncheh, M.; Towhidi, F.; Jamehshorani, S.; Salehiniya, H. Incidence and mortality of prostate cancer and their relationship with the Human Development Index worldwide. Prostate Int. 2016, 4, 118–124.
- WHO. Alcohol, Recorded Per Capita (15+) Consumption (in Litres of Pure Alcohol). Available online: https://www.who.int/data/gho/data/indicators/indicator-details/GHO/alcohol-recorded-per-capita-(15-)-consumption-(in-litres-of-pure-alcohol) (accessed on 19 February 2022).
- Colli, J.L.; Colli, A. International comparisons of prostate cancer mortality rates with dietary practices and sunlight levels. Urol. Oncol. Semin. Orig. Investig. 2006, 24, 184–194.
- Brunner, C.; Davies, N.M.; Martin, R.M.; Eeles, R.; Easton, D.; Kote-Jarai, Z.; Al Olama, A.A.; Benlloch, S.; Muir, K.; Giles, G.; et al. Alcohol consumption and prostate cancer incidence and progression: A Mendelian randomisation study. Int. J. Cancer 2016, 140, 75–85.
- Lin, H.-Y.; Wang, X.; Tseng, T.-S.; Kao, Y.-H.; Fang, Z.; Molina, P.E.; Cheng, C.-H.; Berglund, A.E.; Eeles, R.A.; Muir, K.R.; et al. Alcohol Intake and Alcohol–SNP Interactions Associated with Prostate Cancer Aggressiveness. J. Clin. Med. 2021, 10, 553.
- Ma, L.; Zhao, J.; Li, T.; He, Y.; Wang, J.; Xie, L.; Qin, X.; Li, S. Association between Tumor necrosis factor-alpha gene polymorphisms and prostate cancer risk: A meta-analysis. Diagn. Pathol. 2014, 9, 74.
- Shao, N.; Xu, B.; Mi, Y.-Y.; Hua, L.-X. IL-10 polymorphisms and prostate cancer risk: A meta-analysis. Prostate Cancer Prostatic Dis. 2011, 14, 129–135.
- Bandil, K.; Singhal, P.; Dogra, A.; Bharadwaj, M.; Rawal, S.K.; Doval, D.C.; Varshney, A.K. Association of SNPs/haplotypes in promoter of TNF A and IL-10 gene together with life style factors in prostate cancer progression in Indian population. Agents Actions 2017, 66, 1085–1097.
- Shepard, B.D.; Fernandez, D.J.; Tuma, P.L. Alcohol consumption impairs hepatic protein trafficking: Mechanisms and consequences. Genes Nutr. 2009, 5, 129–140.
- Renau-Piqueras, J.; Gómez-Perretta, C.; Guerri, C.; Sanchis, R. Qualitative and quantitative ultrastructural alterations in hepatocytes of rats prenatally exposed to ethanol with special reference to mitochondria, golgi apparatus and peroxisomes. Virchows Arch. 1985, 405, 237–251.
- Petrosyan, A.; Cheng, P.-W.; Clemens, D.L.; Casey, C.A. Downregulation of the small GTPase SAR1A: A key event underlying alcohol-induced Golgi fragmentation in hepatocytes. Sci. Rep. 2015, 5, 17127.
- Romero, A.M.; Renau-Piqueras, J.; Marín, M.P.; Esteban-Pretel, G. Chronic Alcohol Exposure Affects the Cell Components Involved in Membrane Traffic in Neuronal Dendrites. Neurotox. Res. 2014, 27, 43–54.
- Renau-Piqueras, J.; Miragall, F.; Guerri, C.; Baguena-Cervellera, R. Prenatal exposure to alcohol alters the Golgi apparatus of newborn rat hepatocytes: A cytochemical study. J. Histochem. Cytochem. 1987, 35, 221–228.
- Pandol, S.J.; Gorelick, F.; Gerloff, A.; Lugea, A. Alcohol Abuse, Endoplasmic Reticulum Stress and Pancreatitis. Dig. Dis. 2010, 28, 776–782.
- Ji, C. Mechanisms of Alcohol-Induced Endoplasmic Reticulum Stress and Organ Injuries. Biochem. Res. Int. 2011, 2012, 216450.
- Török, N.; Marks, D.; Hsiao, K.; Oswald, B.J.; McNiven, M.A. Vesicle movement in rat hepatocytes is reduced by ethanol exposure: Alterations in microtubule-based motor enzymes. Gastroenterology 1997, 113, 1938–1948.
- Ren, J.-C.; Zhu, Q.; LaPaglia, N.; Emanuele, N.V.; Emanuele, M.A. Ethanol-induced alterations in Rab proteins: Possible implications for pituitary dysfunction. Alcohol 2005, 35, 103–112.
- Rasineni, K.; McVicker, B.L.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Rab GTPases Associate with Isolated Lipid Droplets (LDs) and Show Altered Content After Ethanol Administration: Potential Role in Alcohol-Impaired LD Metabolism. Alcohol. Clin. Exp. Res. 2013, 38, 327–335.
- Casey, C.A.; Macke, A.J.; Gough, R.R.; Pachikov, A.N.; Morris, M.E.; Thomes, P.G.; Kubik, J.L.; Holzapfel, M.S.; Petrosyan, A. Alcohol-Induced Liver Injury: Down-regulation and Redistribution of Rab3D Results in Atypical Protein Trafficking. Hepatol. Commun. 2021, 6, 374–388.
- Petrosyan, A.; Casey, C.A.; Cheng, P.-W. The role of Rab6a and phosphorylation of non-muscle myosin IIA tailpiece in alcohol-induced Golgi disorganization. Sci. Rep. 2016, 6, 31962.
- Tomás, M.; Marín, M.P.; Martínez-Alonso, E.; Esteban-Pretel, G.; Díaz-Ruiz, A.; Vázquez-Martínez, R.; Malagón, M.M.; Renau-Piqueras, J.; Martínez-Menárguez, J.A. Alcohol induces Golgi fragmentation in differentiated PC12 cells by deregulating Rab1-dependent ER-to-Golgi transport. Histochem. Cell Biol. 2012, 138, 489–501.
- Li, Y.; Ding, W. Impaired Rab7 and dynamin2 block fat turnover by autophagy in alcoholic fatty livers. Hepatol. Commun. 2017, 1, 473–476.
- Tomás, M.; Marín, P.; Megías, L.; Egea, G.; Renau-Piqueras, J. Ethanol perturbs the secretory pathway in astrocytes. Neurobiol. Dis. 2005, 20, 773–784.
- Marmillot, P.; Rao, M.N.; Lakshman, M. Chronic ethanol exposure in rats affects rabs-dependent hepatic trafficking of apolipoprotein E and transferrin. Alcohol 2001, 25, 195–200.
- Larkin, J.M.; Oswald, B.; McNiven, M.A. Ethanol-induced retention of nascent proteins in rat hepatocytes is accompanied by altered distribution of the small GTP-binding protein rab2. J. Clin. Investig. 1996, 98, 2146–2157.
- Casey, C.A.; Bhat, G.; Holzapfel, M.S.; Petrosyan, A. Study of Ethanol-Induced Golgi Disorganization Reveals the Potential Mechanism of Alcohol-Impaired N-Glycosylation. Alcohol. Clin. Exp. Res. 2016, 40, 2573–2590.
- Frisbie, C.P.; Lushnikov, A.Y.; Krasnoslobodtsev, A.V.; Riethoven, J.-J.M.; Clarke, J.L.; Stepchenkova, E.I.; Petrosyan, A. Post-ER Stress Biogenesis of Golgi Is Governed by Giantin. Cells 2019, 8, 1631.
- Petrosyan, A.; Ali, M.F.; Cheng, P.-W. Glycosyltransferase-specific Golgi-targeting Mechanisms. J. Biol. Chem. 2012, 287, 37621–37627.
- Petrosyan, A.; Holzapfel, M.S.; Muirhead, D.E.; Cheng, P.-W. Restoration of Compact Golgi Morphology in Advanced Prostate Cancer Enhances Susceptibility to Galectin-1–Induced Apoptosis by Modifying Mucin O-Glycan Synthesis. Mol. Cancer Res. 2014, 12, 1704–1716.
- Petrosyan, A. Unlocking golgi: Why does morphology matter? Biochemistry 2019, 84, 1490–1501.
- Egea, G.; Franci, C.; Gambus, G.; Lesuffleur, T.; Zweibaum, A.; Real, F.X. cis-Golgi resident proteins and O-glycans are abnormally compartmentalized in the RER of colon cancer cells. J. Cell Sci. 1993, 105, 819–830.
- Kellokumpu, S.; Sormunen, R.; Kellokumpu, I. Abnormal glycosylation and altered Golgi structure in colorectal cancer: Dependence on intra-Golgi pH. FEBS Lett. 2002, 516, 217–224.
- McKinnon, C.M.; Mellor, H. The tumor suppressor RhoBTB1 controls Golgi integrity and breast cancer cell invasion through METTL7B. BMC Cancer 2017, 17, 145.
- Tan, X.; Banerjee, P.; Guo, H.-F.; Ireland, S.; Pankova, D.; Ahn, Y.-H.; Nikolaidis, I.M.; Liu, X.; Zhao, Y.; Xue, Y.; et al. Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. Investig. 2016, 127, 117–131.
- Petrosyan, A. Onco-golgi: Is fragmentation a gate to cancer progression? Biochem. Mol. Biol. J. 2015, 1, 36.
- Manca, S.; Frisbie, C.P.; Lagrange, C.A.; Casey, C.A.; Riethoven, J.-J.M.; Petrosyan, A. The Role of Alcohol-Induced Golgi Fragmentation for Androgen Receptor Signaling in Prostate Cancer. Mol. Cancer Res. 2018, 17, 225–237.
- Kubyshkin, A.V.; Fomochkina, I.I.; Petrosyan, A.M. The impact of alcohol on pro-metastatic n-glycosylation in prostate cancer. Krim Z Eksp Klin Med. 2018, 8, 11–20.
- Stewart, T.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 2502–2511.
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218.
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated Endoplasmic Reticulum Stress Responses in Cancer: Figure 1. Cancer Res. 2007, 67, 10631–10634.
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946.
- Yadav, R.K.; Chae, S.-W.; Kim, H.-R.; Chae, H.J. Endoplasmic Reticulum Stress and Cancer. J. Cancer Prev. 2014, 19, 75–88.
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566.
- Hung, J.H.; Su, I.J.; Lei, H.Y.; Wang, H.C.; Lin, W.C.; Chang, W.T.; Huang, W.; Chang, W.C.; Chang, Y.S.; Chen, C.C.; et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of nf-kappab and pp38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 46384–46392.
- Katanasaka, Y.; Ishii, T.; Asai, T.; Naitou, H.; Maeda, N.; Koizumi, F.; Miyagawa, S.; Ohashi, N.; Oku, N. Cancer antineovascular therapy with liposome drug delivery systems targeted to BiP/GRP78. Int. J. Cancer 2010, 127, 2685–2698.
- Storm, M.; Sheng, X.; Arnoldussen, Y.J.; Saatcioglu, F. Prostate cancer and the unfolded protein response. Oncotarget 2016, 7, 54051–54066.
- Guan, M.; Su, L.; Yuan, Y.-C.; Li, H.; Chow, W.A. Nelfinavir and Nelfinavir Analogs Block Site-2 Protease Cleavage to Inhibit Castration-Resistant Prostate Cancer. Sci. Rep. 2015, 5, 9698.
- Sreenath, T.L.; Macalindong, S.; Mikhalkevich, N.; Sharad, S.; Mohamed, A.; Young, D.; Borbiev, T.; Xavier, C.; Gupta, R.; Jamal, M.; et al. ETS Related Gene mediated Androgen Receptor Aggregation and Endoplasmic Reticulum Stress in Prostate Cancer Development. Sci. Rep. 2017, 7, 1109.
- Mahadevan, N.R.; Rodvold, J.; Almanza, G.; Perez, A.F.; Wheeler, M.C.; Zanetti, M. Er stress drives lipocalin 2 upregulation in prostate cancer cells in an nf-kappab-dependent manner. BMC Cancer 2011, 11, 229.
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111.
- Pachikov, A.N.; Gough, R.R.; Christy, C.E.; Morris, M.E.; Casey, C.A.; LaGrange, C.A.; Bhat, G.; Kubyshkin, A.V.; Fomochkina, I.I.; Zyablitskaya, E.Y.; et al. The non-canonical mechanism of ER stress-mediated progression of prostate cancer. J. Exp. Clin. Cancer Res. 2021, 40, 289.
- Morel, K.L.; Ormsby, R.; Solly, E.L.; Tran, L.N.K.; Sweeney, C.J.; Klebe, S.; Cordes, N.; Sykes, P.J. Chronic low dose ethanol induces an aggressive metastatic phenotype in TRAMP mice, which is counteracted by parthenolide. Clin. Exp. Metastasis 2018, 35, 649–661.
- Wilson, K.M.; Giovannucci, E.L.; Mucci, L.A. Lifestyle and dietary factors in the prevention of lethal prostate cancer. Asian J. Androl. 2012, 14, 365–374.
- Freedland, S.J.; Aronson, W.J. Examining the relationship between obesity and prostate cancer. Rev. Urol. 2004, 6, 73–81.
- Makarem, N.; Lin, Y.; Bandera, E.; Jacques, P.F.; Parekh, N. Concordance with World Cancer Research Fund/American Institute for Cancer Research (WCRF/AICR) guidelines for cancer prevention and obesity-related cancer risk in the Framingham Offspring cohort (1991–2008). Cancer Causes Control 2015, 26, 277–286.
- Graffouillère, L.; Deschasaux, M.; Mariotti, F.; Neufcourt, L.; Shivappa, N.; Hébert, J.R.; Wirth, M.D.; Latino-Martel, P.; Hercberg, S.; Galan, P.; et al. The Dietary Inflammatory Index Is Associated with Prostate Cancer Risk in French Middle-Aged Adults in a Prospective Study. J. Nutr. 2015, 146, 785–791.
- Chhim, A.-S.; Fassier, P.; Latino-Martel, P.; Druesne-Pecollo, N.; Zelek, L.; Duverger, L.; Hercberg, S.; Galan, P.; Deschasaux, M.; Touvier, M. Prospective association between alcohol intake and hormone-dependent cancer risk: Modulation by dietary fiber intake. Am. J. Clin. Nutr. 2015, 102, 182–189.
- Kobayashi, L.C.; Limburg, H.; Miao, Q.; Woolcott, C.; Bedard, L.L.; Massey, T.E.; Aronson, K.J. Folate intake, alcohol consumption, and the methylenetetrahydrofolate reductase (MTHFR) C677T gene polymorphism: Influence on prostate cancer risk and interactions. Front. Oncol. 2012, 2, 100.
- Kenfield, S.A.; Dupre, N.; Richman, E.L.; Stampfer, M.J.; Chan, J.M.; Giovannucci, E.L. Mediterranean Diet and Prostate Cancer Risk and Mortality in the Health Professionals Follow-up Study. Eur. Urol. 2014, 65, 887–894.
- Richard, A.; Faeh, D.; Rohrmann, S.; Braun, J.; Tarnutzer, S.; Bopp, M. Italianity is associated with lower risk of prostate cancer mortality in Switzerland. Cancer Causes Control 2014, 25, 1523–1529.
- Schneider, L.; Su, L.J.; Arab, L.; Bensen, J.T.; Farnan, L.; Fontham, E.T.H.; Song, L.; Hussey, J.; Merchant, A.T.; Mohler, J.L.; et al. Dietary patterns based on the Mediterranean diet and DASH diet are inversely associated with high aggressive prostate cancer in PCaP. Ann. Epidemiol. 2019, 29, 16–22.e1.
- Sharpe, C.R.; Siemiatycki, J. Case-control study of alcohol consumption and prostate cancer risk in Montreal, Canada. Cancer Causes Control 2001, 12, 589–598.
- Hayes, R.B.; Brown, L.M.; Schoenberg, J.B.; Greenberg, R.S.; Silverman, D.T.; Schwartz, A.G.; Swanson, G.M.; Benichou, J.; Liff, J.M.; Hoover, R.N.; et al. Alcohol Use and Prostate Cancer Risk in US Blacks and Whites. Am. J. Epidemiol. 1996, 143, 692–697.
- McGregor, S.E.; Courneya, K.S.; Kopciuk, K.A.; Tosevski, C.; Friedenreich, C.M. Case–control study of lifetime alcohol intake and prostate cancer risk. Cancer Causes Control 2012, 24, 451–461.
- Benedetti, A.; Parent, M.-E.; Siemiatycki, J. Lifetime consumption of alcoholic beverages and risk of 13 types of cancer in men: Results from a case–control study in Montreal. Cancer Detect. Prev. 2009, 32, 352–362.
- Azevedo e Silva, G.; de Rezende, L.F.; Gomes Fda, S.; de Souza Junior, P.R.; Szwarcwald, C.L.; Eluf Neto, J. Lifestyle among former cancer patients in brazil in 2013. Cien. Saude Colet. 2016, 21, 379–388.
- de Menezes, R.F.; Bergmann, A.; de Aguiar, S.S.; Thuler, L.C. Alcohol consumption and the risk of cancer in brazil: A study involving 203,506 cancer patients. Alcohol 2015, 49, 747–751.
- Fillmore, K.M.; Chikritzhs, T.; Stockwell, T.; Bostrom, A.; Pascal, R. Alcohol use and prostate cancer: A meta-analysis. Mol. Nutr. Food Res. 2009, 53, 240–255.
More