Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alexandre Harlé and Version 2 by Amina Yu.
Decoding tumour heterogeneity is a major clinical challenge, considering that it immensely contributes to cancer progression, treatment failure and emergence of drug resistance. Emerging technical and sampling strategies have been developed in order to deeply characterise tumour complexity and clonal architecture, including single-cell profiling, multi-region sampling, representative sampling and longitudinal analysis of liquid biopsy samples.
- tumour heterogeneity
- circulating tumour DNA
- liquid biopsy
- next-generation sequencing
- multi-region sampling
- single-cell approaches
- treatment resistance
1. Tumour Heterogeneity: From Historical Perspectives to Novel Insights
1.1. Varying Degrees of Tumour Heterogeneity
Tumour heterogeneity harbours multiple layers of complexity in human malignancies. It has long been known that tumours of the same histopathological subtype commonly differ from one patient to another (inter-tumour heterogeneity) (Figure 1). Exacerbating the complexity even further, sizable variations have been reported within a single tumour (intra-tumour heterogeneity, ITH). ITH can be detected between the different geographic regions of the same primary tumour or even between the primary tumour and the metastastic lesions (spatial intra-tumour heterogeneity). Moreover, the analysis of serial tumour samples demonstrated that the cell features may evolve during the course of the disease progression (temporal heterogeneity) under environmental or therapeutic stress [1][2][1,2]. ITH has been observed in most (nearly all) types of cancers, including both haematological malignancies (chronic lymphoblastic leukemia and acute lymphoblastic leukemia), and solid tumours (lung, breast, ovarian, pancreatic, kidney, colorectal, brain and prostate cancers) [3].
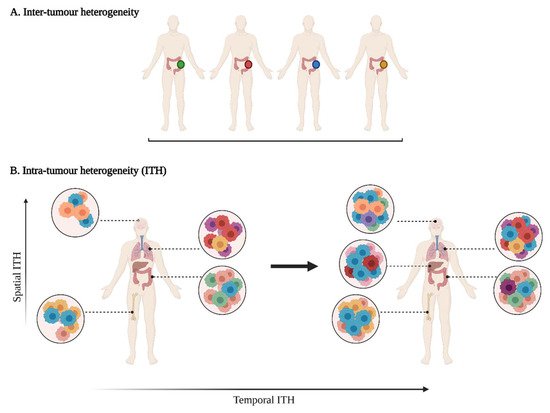
Figure 1. A multifaceted heterogeneity in cancers. (A) Inter-tumour heterogeneity refers to the variability observed in tumours of the same histological subtypes between different patients. (B) Intra-tumour heterogeneity (ITH) is observed across different regions of the primary tumour site and/or metastatic sites (spatial ITH) and can evolve over time (temporal ITH). Colours represent the different characteristics between tumours or tumour cells.
1.1.1. Phenotypic Heterogeneity
The first demonstration of tumour heterogeneity has been made by histopathologists who are familiar with morphological divergence (differentiation status, necrosis, fibrosis, and so onetc.) across the tumours or between the different areas of the tumour (Figure 2) [1][4][5][6][7][8][1,4,5,6,7,8]. This notion has led to the very basis of tumour classification systems based on histopathological features [9]. Tumour grading systems notably include the pathological examination of multiple microscopy fields in order to avoid tumour misclassification due to ITH [10]. Increasing evidence indicates that tumour foci are heterogeneous at other phenotypic levels than merely morphologic, including differential capabilities in terms of proliferation, metabolism, motility, migration, invasiveness, metastasis and stemness, as well as varied sensitivity to therapies [11][12][13][11,12,13]. The morphological and other phenotypic cell features co-vary in the different tumour regions, notably between the core and the external borders of the tumour.
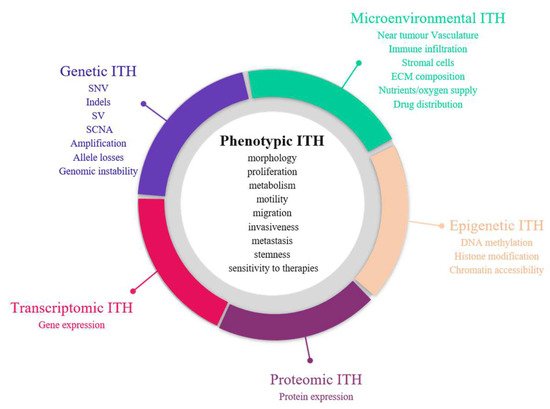
Figure 2. Sources of intra-tumour heterogeneity (ITH). Genetic, epigenetic, microenvironmental, transcriptomic and proteomic heterogeneities coexist in tumours and are linked with phenotypic diversity. Abbreviations: CAF: Cancer-associated fibroblasts; ECM: extracellular matrix; EMT: epithelial-to-mesenchymal transition; indels: small insertions and deletions; LOH: loss of heterozygoty; MET: mesenchymal-to-epithelial transition; SCNA: somatic copy number alterations; SNV: single nucleotide variants; SV: structural variants.
1.1.2. Molecular Heterogeneity
Advances in next-generation sequencing (NGS) revealed the extent and prevalence of molecular diversity in tumours [14][15][14,15]. The sequencing of multiple regions in space and time demonstrated the various repertoires of genetic events that can occur in cancers, including single nucleotide variants (SNVs), small insertions and deletions (indels), structural variants and somatic copy number alterations (SCNA) [15]. Large-scale studies indicated that genetic ITH occurs in almost all cancer types, albeit at varying degrees [16][17][16,17]. Melanoma and lung adenocarcinomas notably account for cancers with high mutational tumour burden and the establishment of specific mutational signatures as a result of exposure to exogenous mutagens (UV radiation and tobacco smoke) [18]. Dietz et al. demonstrated that the frequencies of driver gene mutations in regionally distinct areas of lung adenocarcinomas were correlated with the spatial distribution of histological patterns, highlighting an interplay between histologic and genetic features in a tumour [19][20][19,20].
However, the genetic perspective is insufficient to fully explain the range of phenotypic diversity in solid malignancies, given the fact that cell populations with identical genetic background can lead to distinct morphological patterns and differential responses to treatment or environmental stimuli [10][21][10,21]. Emerging evidence demonstrated that ITH also take place at other levels, such as epigenetics, transcriptomics and proteomics [22].
The epigenome is defined as a connection between the genome and the environment. Alterations of the epigenetic machinery has been recognised as a hallmark of cancer [23] and may appear early during carcinogenesis [24]. Epigenetic marks induce heritable changes in gene expression without any modification in the underlying DNA sequence that allows cells to adapt to microenvironment stimuli (oxygen, nutrient deprivation, acidity, and so onetc.) and develop resistance mechanisms against anticancer therapies [25]. Bidirectional communications between genetics and epigenetics have been reported in cancers, with the detection of somatic mutations in genes encoding epigenome regulators (such as DNMT3A, IDH1, H3F3A) and inversely the identification of DNA hypomethylation or epigenetic silencing of DNA repair genes (such as MLH1 or BRCA1) that can cause genomic instability in cancer cells [24]. It waStudies assessing histone modifications, chromatin accessibility and DNA methylation profiles demonstrated a high epigenetic variability in cancers [21][26][27][21,26,27]. Considering the major implications of epigenetics in the development of cancers and their response to anticancer treatments, a better understanding of epigenetic heterogeneity could help to identify novel epigenetic therapies and consider them for a combination with other anticancer treatments (genotoxic/cytotoxic agents, hormone therapy, immunotherapy, targeted therapy) to improve their efficacy or reverse drug resistance [25].
Transcriptome refers to all RNA species that can be found in cells; however, mRNAs are frequently the most studied. Their composition varies between cell types and tumour types and continuously evolves depending on the local conditions that are applied to cells over time. They can be explored through targeted (RT-PCR) or high throughput approaches (gene expression arrays, RNA sequencing (RNASeq)). A plethora of gene expression signatures have been developed in oncology for tumour classification [28], prognosis establishment [29][30][31][32][29,30,31,32], therapeutic and surveillance decision making [33] but only a few are already implemented for routine practice [34].
Because proteins directly reveal the functional mechanisms that occur in cancers and account for most of the therapeutic targets, it appears important to assess tumour heterogeneity at the protein level, which has shown growing interest. Proteomic approaches have long lagged behind those for transcriptome and genome due to technical limitations, high amounts of proteins generated from a single gene (with different isoforms and modification states) and a complex regulation of protein expression at both translational and post-translational levels [35]. Immunohistochemistry appears as one of the most standard approaches to assess protein abundance changes; however, it provides only semi-quantitative information, interrogates a limited number of proteins and is limited by the availability of appropriate antibodies. The development of reverse-phase protein array (RPPA) and mass spectrometry (MS)-based methods enabled the assessment of the proteomic landscape on a larger scale [35]. Transcriptomic approaches cannot substitute proteomic investigations, as the analysis of datasets from The Cancer Proteome Atlas (TCPA) found a poor correlation between protein and gene expression in cancer tissues, with Spearman correlation oscillating from 0.1 to 0.3, depending on the cancer type [36].
Recognition of all these cancer-specific molecular processes as major elements in the evolution of cancers and their considerable diversity has led to the launch of cancer genomics programs by international consortia, such as The Cancer Genome Atlas consortium (TCGA) and the International Cancer Genome Consortium (ICGC). All aim to integrate the analysis of multi-omics datasets (genetic, epigenetic, transcriptomic and proteomic data) to provide a comprehensive overview of the tumour landscape [16][37][38][39][40][41][42][43][44][45][16,37,38,39,40,41,42,43,44,45].
1.1.3. Tumour Micro-Environment (TME) Heterogeneity
The influence of the complex ecosystem in which cancer cells evolve has long been overlooked. In the last decade, cancer biology progressively shifted from a cancer cell-centric model to a more ample view, where cancer cells and their near environment are highly interrelated. The tumour microenvironment (TME) is made up of non-transformed cells (endothelial cells, fibroblasts, pericytes, adipocytes, immune cells, and so onetc.) and non-cellular constituents (such as the extracellular matrix) which are shaped by cancer cells through the modification of local environmental conditions and the secretion of oncogenic signals [46][47][46,47]. As a consequence, the phenotypic traits and behaviours of TME components are highly heterogeneous, depending on the tumour context [46][48][46,48]. In return, TME can assist in the development of the tumour niche by contributing to cancer progression, metastasis and drug resistance [48][49][50][48,49,50]. In this way, TME represents an emerging target for treatments (such as immune checkpoint inhibitors or antiangiogenic therapies) and should be taken into consideration for clinical decisions. Recently, Garattini et al. demonstrated that heterogeneity also extends to the drug distribution in tumours, which depends on many aspects of the patient, the tumour and its microenvironment and influences tumour response [51].
1.2. Unravelling Evolutionary Processes behind Tumour Heterogeneity
Two major and paradoxical theories have been developed to explain the installation of high degree of diversity in tumours. In 1976, Peter Nowell first described the cancer development as a continuous evolutionary process originating from a single renagade mutant cell and driven by the accumulation of stepwise somatic mutations during proliferation processes that give rise to various clones and subclones [52][61]. The development of multiple cell groups with distinct genomic profiles is amplified by genomic instability that arises in most solid tumours and haematopoietic malignancies as a result from both exposure to exogenous mutagens and defects in DNA repair pathways [53][62]. A subclone is characterised as a set of cells that diverge from the cell ancestor lineage (clone) by the presence of additional genetic alterations. Equivalent to Darwinian natural selection, most stochastic events that appears during the evolution process probably do not confer any selective benefit to the cancer cells (passenger mutations) [54][63]. In contrast, certain mutations can provide a fitness advantage over adjacent cells (driver mutations) and enables them to become predominant and outcompete other ones [55][64]. Most driver mutations are clonal. They appear early during cancer progression under a given microenvironmental context and foster cancer progression but they seem not essential for cancer maintenance once installed [54][63]. The clonal genomic architecture is distinct from a tumour to another considering that the emergence of subclones strongly depends on specific environmental stresses (local hypoxia or inflammation, treatment exposure, etc.) applied in each tumour over time. More recently, epigenetics and genetics were shown to follow convergent evolutionary trajectories in the development of cancers, highlighting the potential interest of combining epigenetic agents with other anticancer therapies [56][57][58][65,66,67].
In contrast to the Darwinian clonal evolution theory, where all subclones possess tumorigenic potential, a second model proposed that only a small subgroup of cancer cells (named cancer stem cells (CSC) or tumour-initiating cells) has the capacity to generate new tumours [59][60][68,69]. In this model, tumours are structured in a unidirectional hierarchy fashion, whereby CSC can either indefinitely self-renew (symmetric division) or differentiate into multiple cancer cell types (asymmetric division). CSC with stem cell-like characteristics have been observed in several cancers, including leukemia, breast, colon, head and neck and oesophageal cancers [55][64]. CSC are thought to be more drug-resistant than non-CSC and in such ways, they may be responsible for recurrence and therapeutic evasion [59][68]. Increasing evidence, however, indicated that non-CSC can readily convert to a CSC state through cell plasticity programmes, such as epithelial-to-mesenchymal transition (EMT), indicating that the hierarchy seems less rigid than previously thought [61][70]. In the same manner, different subsets of CSC with variable EMT phenotypes can coexist in tumours and can switch from one to another [61][70]. Stemness and CSC plasticity may be modulated by internal (genetic and epigenetic) and external (TME) factors that can work apart or simultaneously [62][71]. Van Niekerk and colleagues show that certain stem cell features can be acquired by cancer cells through clonal selection, highlighting the fact that clonal evolution and the CSC theories are not necessarily mutually exclusive and can intertwine [63][72].
In these first models of tumour evolution, ITH was thought to gain gradually over time as the tumour grew (Figure 3). Although this concept of continuous clonal evolution is still applicable to describe most cancer evolutionary processes, increasing evidence supported the idea that this model cannot explain the full spectrum of observed evolutionary behaviours [64][73]. Notably, single catastrophic events, such as whole-genome doubling, chromosomal chromoplexy and chromothripsis, can arise suddenly as single macroevolutionary jumps over long periods of relative stasis. In some extreme cases of punctuated tumour evolution, the development of colorectal cancers and other tumour types has been modelled as “Big-bang” dynamics, whereby a single or few mutational bursts occur early during carcinogenesis and result in a large number of intermixed subclones that are not subjected to selective pressure and coexist during growth (neutral evolution) [65][66][67][74,75,76].
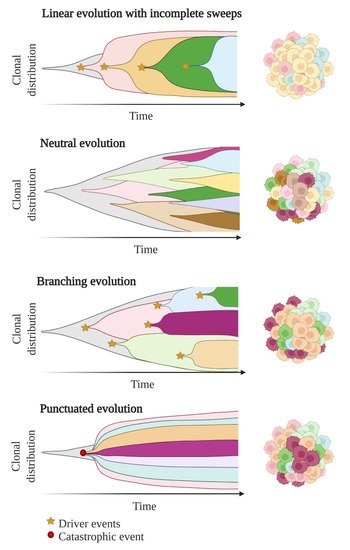
Figure 3. Models of tumour evolution described by Muller plots, which represent the tumour clonal dynamics over time. Colours indicate the different genotypes of the tumour cell clones.
Branched evolutionary trajectories have been extensively described in a wide range of tumour types, such as childhood acute lymphoblastic leukaemia, clear cell renal carcinomas, pancreatic, colorectal, breast and prostate cancers [68][77]. Evolutionary pathways can then be represented as a phylogenetic tree, where truncal mutations (clonal) represent the alterations occurring early in cancer development progenitors, while nontruncal mutations (subclonal) emerge during cancer progression and are shared by only a small group but not all cancer cells. In a branched evolution pattern, several distinct subclones co-exist and can be either intermingled in the same area or regionally separated, depending on the presence of physical barriers, such as blood vessels or microenvironment specificities [53][62].
ITH has also been described in cancer cases with linear evolutionary trajectories whereby a predominant subclone outgrows at the expense of its predecessor(s) followed by incomplete selective sweeps [53][68][62,77]. Although most studies described a single model of evolution in cancers, emerging data suggest that tumours may follow different models of evolution (linear, branched, punctuated or neutral) sequentially or simultaneously during the course of the disease [69][78]. The full context of tumour evolution is still to be explored in detail in order to better define effective therapeutic strategies.
2. Clinical Consequences of Tumour Heterogeneity
2.1. Impact on Diagnosis, Prognosis and Therapeutic Predictions
In the last decades, the management of patients with cancer has been revolutionised by better knowledge on the molecular background in cancer development. The understanding of molecular inter-tumour heterogeneity has formed the basis of personalised medicine in diagnosis, prognosis and treatment of cancers. Notably, it has set the limits of using universal anticancer drugs and has been a major driver for the emergence of novel therapies targeting specific molecular characteristics [70][79]. For most cancers, molecular diagnosis has entered in clinical practice as a prerequisite for tumour subtyping, prognosis refinement and treatment-decision making. Molecular testing is routinely performed on a limited tumour tissue area selected by the pathologist to be the most representative of the tumour. However, such methodology induces inherent under-sampling bias due to spatial and temporal ITH.
2.1.1. Tumour Sampling Bias Due to Spatial ITH
Most of the histopathological and molecular features are not expressed homogeneously in tumour subpopulations, highlighting the fact that the analysis of a single sample may lead to diagnostic and prognostic errors and provide an incomplete view of potential vulnerabilities to treatment [71][80]. The identification of an actionable mutation in a predominant subline might not necessarily predict the response of the bulk tumour [70][79]. If the targeted mutation is shared by only a subset of cancer cells in the tumour (nontruncal mutation), the response to the targeted therapy is often of limited duration due to the outgrowth of resistant pre-existing subclones and/or the development of new drug-tolerant clones under therapeutic selection pressure [72][81]. In patients with metastatic disease, it is of clinical importance to portray ITH, given the fact that a genetic shift is infrequently observed between a metastase and the primary tumour site or even between two spatially distinct metastases [2].
2.1.2. Tumour Sampling Bias Due to Temporal ITH
Archival tissue specimens commonly serve as starting material for testing if any recent sample is available. These samples can be collected many months or years previously, at the time of diagnosis or when a new lesion appears at distance. However, they cannot reliably reflect the tumour landscape over the time, considering that the tumour constantly evolves under specific microenvironmental conditions (such as acidosis, hypoxia or reactive oxygen species) [73][82] or exposure to therapeutic lines (DNA damaging agents or radiotherapy, targeted therapy and immunotherapy) [74][83]. In Tthis context, treatment failure may happen when therapy is directed against a specific molecular characteristic [10].
2.1.3. Determining ITH to Decipher the Identity of the Tumour or of Specific Regions of the Tumour
Analysing the characteristics of a tumour or of different tumour regions allows us to define the tissue of origin but also provide insights into the molecular events that occurred sequentially or in parallel throughout the development of the tumour. E In this context, evaluating ITH gives a remarkable view of the whole history of the tissue and could help to better understand cancerogenesis and develop new therapeutic strategies.
2.1.4. ITH Is Associated with Poorer Clinical Outcomes
The analysis of data collected by the TCGA from more than 3300 tumours across nine tumour types revealed that ITH has prognostic utility [75][84]. High degree of ITH are closely related to poorer immune infiltration and worse prognosis for patients with solid malignancies, including head and neck carcinomas, glioma, melanomas urothelial, breast, renal, lung and prostate cancers [75][84]. The relationship between ITH and patient outcomes is, however, complex to interpret and can be influenced by many aspects of the tumour, including the tumour cell of origin, the number of clones, the level of chromosomal instability, the type of somatic events and their order of appearance [76][85]. The prognostic value of ITH concerns other aspects than just genetic diversity [77][78][79][80][86,87,88,89], suggesting the importance of capturing the full extent of ITH.
2.1.5. High Amounts of Biomarkers to Analyse in Order to Fully Decipher ITH
Emerging omics technologies have shown their interest in recent studies to analyse all types of ITH. However, multi-omics analyses are still far from standard-of-care, considering their cost, their low spread in clinical labs and the need for powerful data storage options for these big data. The analysis of only few histomolecular biomarkers in tumours is still the reference and may lead to misinterpretation.
Considering the huge amounts of data generated from multi-omics approaches and their high complexity, there is a considerable need to develop automation tools able to provide an integrative analysis of the multiple layers of heterogeneity without any expert intervention. Last advances in artificial intelligence and machine learning models allowed us to better predict cell subtypes and infer their proportions in tumours and TME based on their inherent multi-omics characteristics. These approaches have the potential to integrate both molecular and histopathological imaging data to refine tumour heterogeneity in the spatial context and go beyond what can be distinguished by routine microscopy observations [81][82][90,91]. However, due to their recent development, they still lack standardisation and need further evaluation prior to their implementation in a clinical setting [82][91].
2.2. Impact on Therapeutic Strategies
Although the notion of ITH and its impact on therapeutic response is now well documented in research studies, ITH determination is rarely taken into account in current clinical decision making that mostly relies on short-term treatment efficacy and the detection of resistance mechanisms to adapt the treatment and forestall disease relapse. However, capturing ITH could aid in developing novel strategies to provide long-term drug response and minimise the emergence of resistance mechanisms [74][83]. For example, in cases of heterogeneous tumours, upfront combination of therapies targeting different cancer cell subpopulations or dependencies could help to obtain a more durable response by minimising ITH and hindering minor subclones to expand under monotherapy pressure. Given the high molecular diversity that can be observed between the different regions of tumours, targeting all alterations is clearly unrealistic in clinical practice. The development of such strategies would require the determination of the aberrations the most critical for cellular functions and survival beforehand. The use of targeted therapy associated with non-targeted agents or ITH-reducing agents (such as histone deacetylase (HDACi), bromodomain and extra-terminal protein (BETi) or histone demethylase (HDMi) inhibitors) could also be considered to prevent the emergence of resistance in the case of highly heterogeneous distribution of a molecular target in tumour tissues [74][83]. Some groups also proposed adaptive therapies as a way to stabilise the balance between drug-sensitive and drug-resistant subclones and maintain tumour burden [83][84][85][92,93,94].