The proteolytical cleavage of transmembrane proteins with subsequent release of their extracellular domain, so-called ectodomain shedding, is a post-translational modification that plays an essential role in several biological processes, such as cell communication, adhesion and migration. Metalloproteases are major proteases in ectodomain shedding, especially the disintegrin metalloproteases (ADAMs) and the membrane-type matrix metalloproteases (MT-MMPs), which are considered to be canonical sheddases for their membrane-anchored topology and for the large number of proteins that they can release. The unique ability of TIMP-3 to inhibit different families of metalloproteases, including the canonical sheddases (ADAMs and MT-MMPs), renders it a master regulator of ectodomain shedding.
- TIMPs
- ADAMs
- metalloproteases
- ectodomain shedding
- proteomics
1. Introduction
2. Regulation of TIMP-3
Given its role in modulating ECM turnover and ectodomain shedding, the bioavailability of TIMP-3 must be finely regulated. This occurs at several levels, including transcriptional regulation, by growth factors and cytokines, and epigenetically by promoter methylation, post-transcriptional regulation by specific miRNAs and by receptor-mediated endocytosis (Figure 1).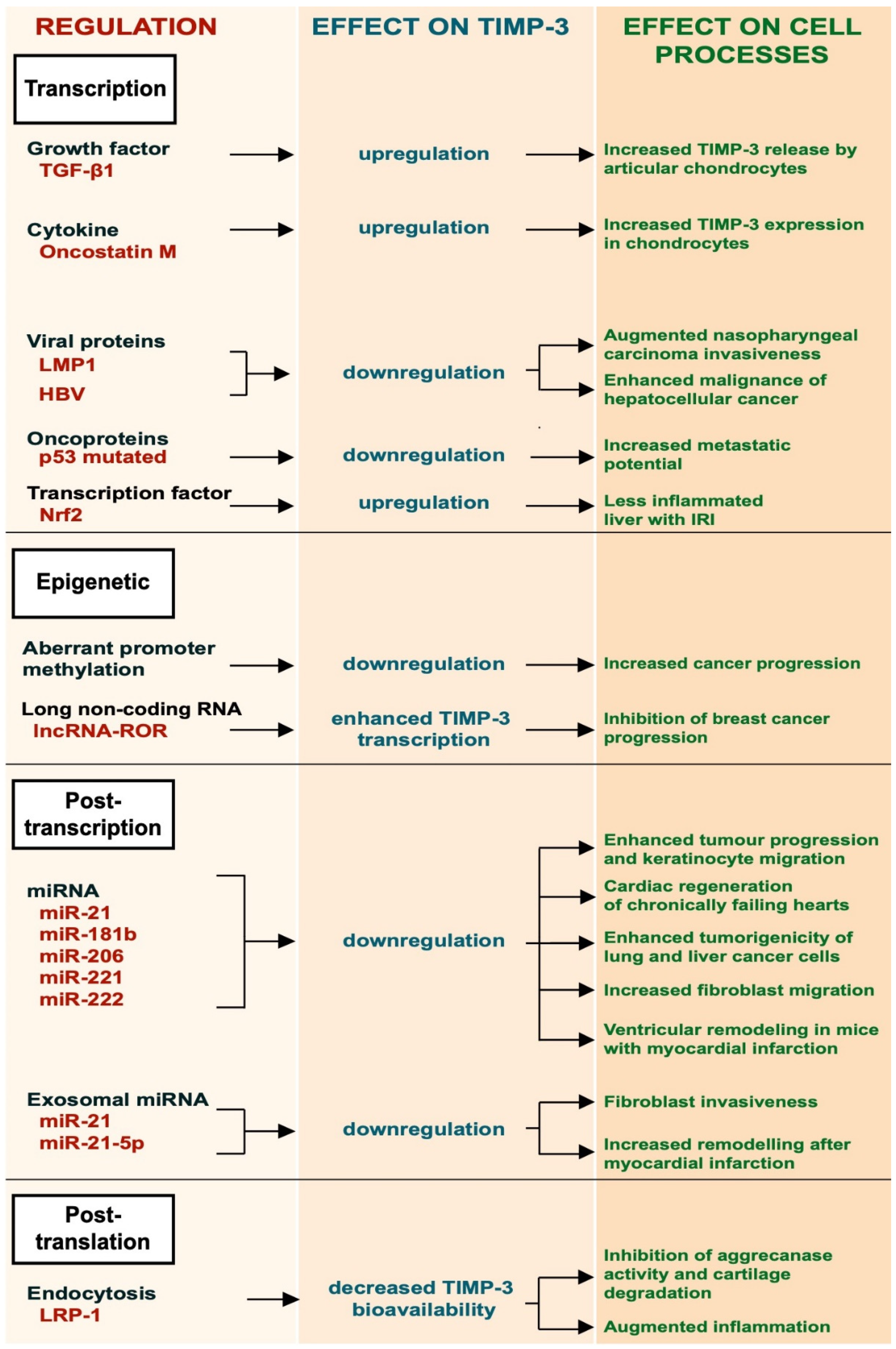 Figure 1. Schematic representation of the different regulatory mechanisms of TIMP-3.
Figure 1. Schematic representation of the different regulatory mechanisms of TIMP-3.2.1. Transcriptional Regulation
2.2. Epigenetic Regulation
2.3. Post-Transcriptional Regulation
2.4. LRP-1-Mediated Endocytosis
3. Mass Spectrometry-Based Approaches to Investigate Functions of TIMP-3
The function of a protease strictly depends on the collection of substrates that it cleaves. Some proteases cleave a large number of substrates, while others have a very restricted activity towards a few proteins. Furthermore, the activity of proteases can be spatially and/or temporally confined, so that even proteases with a large spectrum of substrates may only access a limited number of them at specific locations and at specific times. In the last years, high-resolution mass spectrometry techniques have been developed to investigate the function of specific proteases by the systematic identification of their substrates in a specific cellular case [2][37]. These proteomics-based approaches have also been applied to TIMP-3, enabling the discovery of new functions of the inhibitor on the stabilization of transmembrane proteins and endocytosis of secreted proteins [38][39]. Here will provide a brief description of these mass spectrometry-based techniques that have been used to characterize the function of TIMP-3 and its target proteases and which may eventually be applied to other proteases and their inhibitors.3.1. High-Resolution Secretome Analysis
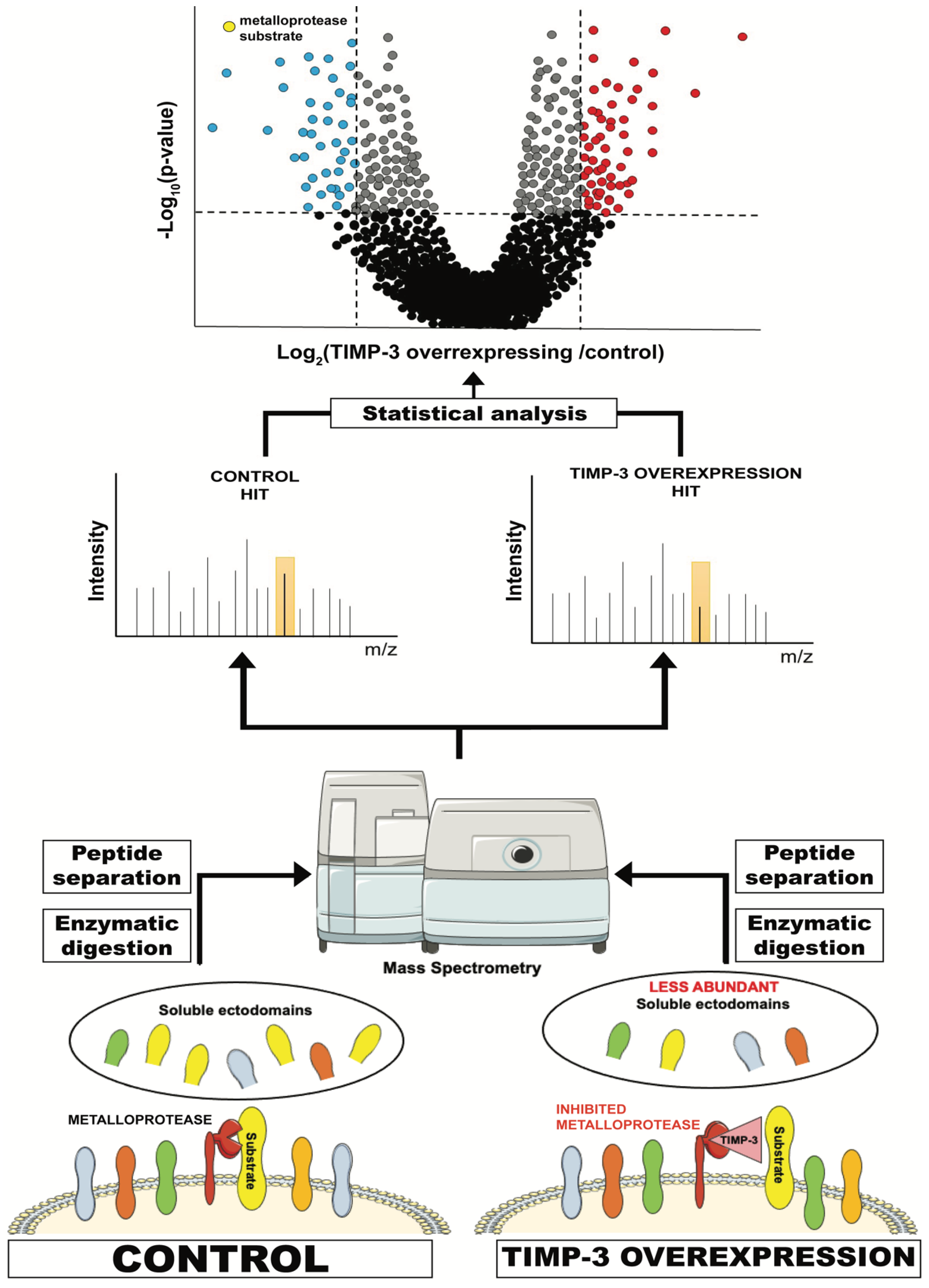 Figure 2. Schematic representation of a typical proteomic workflow to investigate ectodomain shedding. For instance, TIMP-3-overexpressing or control cells are cultured in serum-free media and conditioned media are collected. These contain, among other proteins secreted by the cell, the ectodomain of proteins that are shed by TIMP-3 target metalloproteases. Levels of these proteins in TIMP-3-overexpressing cells will be lower due to inhibition of their shedding by TIMP-3. Conditioned media from TIMP-3-overexpressing and control cells will be applied to tryptic digestion, C18 reversed phase liquid chromatography (LC) and, ultimately, MS/MS analysis. This will enable the identification of proteins contained in the conditioned media and the quantification of their levels in TIMP-overexpressing cells versus controls. Finally, a statistical analysis will show the levels of the proteins that are altered in the media of TIMP-3-overexpressing cells.
Figure 2. Schematic representation of a typical proteomic workflow to investigate ectodomain shedding. For instance, TIMP-3-overexpressing or control cells are cultured in serum-free media and conditioned media are collected. These contain, among other proteins secreted by the cell, the ectodomain of proteins that are shed by TIMP-3 target metalloproteases. Levels of these proteins in TIMP-3-overexpressing cells will be lower due to inhibition of their shedding by TIMP-3. Conditioned media from TIMP-3-overexpressing and control cells will be applied to tryptic digestion, C18 reversed phase liquid chromatography (LC) and, ultimately, MS/MS analysis. This will enable the identification of proteins contained in the conditioned media and the quantification of their levels in TIMP-overexpressing cells versus controls. Finally, a statistical analysis will show the levels of the proteins that are altered in the media of TIMP-3-overexpressing cells.3.2. Surfaceomics
3.3. SPECS and SUSPECS
3.4. TAILS
4. Conclusions and Perspectives
References
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018, 37, e99456.
- Muller, S.A.; Scilabra, S.D.; Lichtenthaler, S.F. Proteomic Substrate Identification for Membrane Proteases in the Brain. Front. Mol. Neurosci. 2016, 9, 96.
- Murphy, G.; Nagase, H. Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J. 2011, 278, 2–15.
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289.
- Fiore, E.; Fusco, C.; Romero, P.; Stamenkovic, I. Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene 2002, 21, 5213–5223.
- Rodriguez-Manzaneque, J.C.; Carpizo, D.; Plaza-Calonge Mdel, C.; Torres-Collado, A.X.; Thai, S.N.; Simons, M.; Horowitz, A.; Iruela-Arispe, M.L. Cleavage of syndecan-4 by ADAMTS1 provokes defects in adhesion. Int. J. Biochem. Cell Biol. 2009, 41, 800–810.
- Scharfenberg, F.; Helbig, A.; Sammel, M.; Benzel, J.; Schlomann, U.; Peters, F.; Wichert, R.; Bettendorff, M.; Schmidt-Arras, D.; Rose-John, S.; et al. Degradome of soluble ADAM10 and ADAM17 metalloproteases. Cell. Mol. Life Sci. 2020, 77, 331–350.
- Hsia, H.E.; Tushaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell. Mol. Life Sci. 2019, 76, 3055–3081.
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71.
- Qureshi, H.Y.; Sylvester, J.; El Mabrouk, M.; Zafarullah, M. TGF-beta-induced expression of tissue inhibitor of metalloproteinases-3 gene in chondrocytes is mediated by extracellular signal-regulated kinase pathway and Sp1 transcription factor. J. Cell. Physiol. 2005, 203, 345–352.
- Qureshi, H.Y.; Ahmad, R.; Sylvester, J.; Zafarullah, M. Requirement of phosphatidylinositol 3-kinase/Akt signaling pathway for regulation of tissue inhibitor of metalloproteinases-3 gene expression by TGF-beta in human chondrocytes. Cell Signal. 2007, 19, 1643–1651.
- Li, W.Q.; Dehnade, F.; Zafarullah, M. Oncostatin M-induced matrix metalloproteinase and tissue inhibitor of metalloproteinase-3 genes expression in chondrocytes requires Janus kinase/STAT signaling pathway. J. Immunol. 2001, 166, 3491–3498.
- Chang, S.H.; Chang, H.C.; Hung, W.C. Transcriptional repression of tissue inhibitor of metalloproteinase-3 by Epstein-Barr virus latent membrane protein 1 enhances invasiveness of nasopharyngeal carcinoma cells. Oral Oncol. 2008, 44, 891–897.
- Kim, J.R.; Kim, C.H. Association of a high activity of matrix metalloproteinase-9 to low levels of tissue inhibitors of metalloproteinase-1 and -3 in human hepatitis B-viral hepatoma cells. Int. J. Biochem. Cell Biol. 2004, 36, 2293–2306.
- Thomas, S.; Reisman, D. Localization of a mutant p53 response element on the tissue inhibitor of metalloproteinase-3 promoter: Mutant p53 activities are distinct from wild-type. Cancer Lett. 2006, 240, 48–59.
- Rao, J.; Qiu, J.; Ni, M.; Wang, H.; Wang, P.; Zhang, L.; Wang, Z.; Liu, M.; Cheng, F.; Wang, X.; et al. Macrophage nuclear factor erythroid 2-related factor 2 deficiency promotes innate immune activation by tissue inhibitor of metalloproteinase 3-mediated RhoA/ROCK pathway in the ischemic liver. Hepatology 2021.
- Bachman, K.E.; Herman, J.G.; Corn, P.G.; Merlo, A.; Costello, J.F.; Cavenee, W.K.; Baylin, S.B.; Graff, J.R. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res. 1999, 59, 798–802.
- Rohrs, S.; Dirks, W.G.; Meyer, C.; Marschalek, R.; Scherr, M.; Slany, R.; Wallace, A.; Drexler, H.G.; Quentmeier, H. Hypomethylation and expression of BEX2, IGSF4 and TIMP3 indicative of MLL translocations in acute myeloid leukemia. Mol. Cancer 2009, 8, 86.
- Eads, C.A.; Lord, R.V.; Wickramasinghe, K.; Long, T.I.; Kurumboor, S.K.; Bernstein, L.; Peters, J.H.; DeMeester, S.R.; DeMeester, T.R.; Skinner, K.A.; et al. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001, 61, 3410–3418.
- Hu, A.; Hong, F.; Li, D.; Jin, Y.; Kon, L.; Xu, Z.; He, H.; Xie, Q. Long non-coding RNA ROR recruits histone transmethylase MLL1 to up-regulate TIMP3 expression and promote breast cancer progression. J. Transl. Med. 2021, 19, 95.
- Gabriely, G.; Wurdinger, T.; Kesari, S.; Esau, C.C.; Burchard, J.; Linsley, P.S.; Krichevsky, A.M. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol. Cell. Biol. 2008, 28, 5369–5380.
- Wang, B.; Hsu, S.H.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene 2010, 29, 1787–1797.
- Song, B.; Wang, C.; Liu, J.; Wang, X.; Lv, L.; Wei, L.; Xie, L.; Zheng, Y.; Song, X. MicroRNA-21 regulates breast cancer invasion partly by targeting tissue inhibitor of metalloproteinase 3 expression. J. Exp. Clin. Cancer Res. 2010, 29, 29.
- Selaru, F.M.; Olaru, A.V.; Kan, T.; David, S.; Cheng, Y.; Mori, Y.; Yang, J.; Paun, B.; Jin, Z.; Agarwal, R.; et al. MicroRNA-21 is overexpressed in human cholangiocarcinoma and regulates programmed cell death 4 and tissue inhibitor of metalloproteinase 3. Hepatology 2009, 49, 1595–1601.
- Yang, X.; Wang, J.; Guo, S.L.; Fan, K.J.; Li, J.; Wang, Y.L.; Teng, Y.; Yang, X. miR-21 promotes keratinocyte migration and re-epithelialization during wound healing. Int. J. Biol. Sci. 2011, 7, 685–690.
- Limana, F.; Esposito, G.; D’Arcangelo, D.; Di Carlo, A.; Romani, S.; Melillo, G.; Mangoni, A.; Bertolami, C.; Pompilio, G.; Germani, A.; et al. HMGB1 attenuates cardiac remodelling in the failing heart via enhanced cardiac regeneration and miR-206-mediated inhibition of TIMP-3. PLoS ONE 2011, 6, e19845.
- Garofalo, M.; Di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 2009, 16, 498–509.
- Wang, C.; Wang, Y.; Chang, X.; Ba, X.; Hu, N.; Liu, Q.; Fang, L.; Wang, Z. Melanoma-Derived Exosomes Endow Fibroblasts with an Invasive Potential via miR-21 Target Signaling Pathway. Cancer Manag. Res. 2020, 12, 12965–12974.
- Dong, J.; Zhu, W.; Wan, D. Downregulation of microRNA-21-5p from macrophages-derived exosomes represses ventricular remodeling after myocardial infarction via inhibiting tissue inhibitors of metalloproteinase 3. Int. Immunopharmacol. 2021, 96, 107611.
- Scilabra, S.D.; Troeberg, L.; Yamamoto, K.; Emonard, H.; Thogersen, I.; Enghild, J.J.; Strickland, D.K.; Nagase, H. Differential regulation of extracellular tissue inhibitor of metalloproteinases-3 levels by cell membrane-bound and shed low density lipoprotein receptor-related protein 1. J. Biol. Chem. 2013, 288, 332–342.
- Troeberg, L.; Fushimi, K.; Khokha, R.; Emonard, H.; Ghosh, P.; Nagase, H. Calcium pentosan polysulfate is a multifaceted exosite inhibitor of aggrecanases. FASEB J. 2008, 22, 3515–3524.
- Carreca, A.P.; Pravata, V.M.; Markham, M.; Bonelli, S.; Murphy, G.; Nagase, H.; Troeberg, L.; Scilabra, S.D. TIMP-3 facilitates binding of target metalloproteinases to the endocytic receptor LRP-1 and promotes scavenging of MMP-1. Sci. Rep. 2020, 10, 12067.
- Scilabra, S.D.; Yamamoto, K.; Pigoni, M.; Sakamoto, K.; Muller, S.A.; Papadopoulou, A.; Lichtenthaler, S.F.; Troeberg, L.; Nagase, H.; Kadomatsu, K. Dissecting the interaction between tissue inhibitor of metalloproteinases-3 (TIMP-3) and low density lipoprotein receptor-related protein-1 (LRP-1): Development of a “TRAP” to increase levels of TIMP-3 in the tissue. Matrix Biol. 2017, 59, 69–79.
- Liu, Q.; Zhang, J.; Tran, H.; Verbeek, M.M.; Reiss, K.; Estus, S.; Bu, G. LRP1 shedding in human brain: Roles of ADAM10 and ADAM17. Mol. Neurodegener 2009, 4, 17.
- Yamamoto, K.; Santamaria, S.; Botkjaer, K.A.; Dudhia, J.; Troeberg, L.; Itoh, Y.; Murphy, G.; Nagase, H. Inhibition of Shedding of Low-Density Lipoprotein Receptor-Related Protein 1 Reverses Cartilage Matrix Degradation in Osteoarthritis. Arthritis Rheumatol. 2017, 69, 1246–1256.
- Schubert, K.; Collins, L.E.; Green, P.; Nagase, H.; Troeberg, L. LRP1 Controls TNF Release via the TIMP-3/ADAM17 Axis in Endotoxin-Activated Macrophages. J. Immunol. 2019, 202, 1501–1509.
- Yang, C.Y.; Troeberg, L.; Scilabra, S.D. Quantitative Mass Spectrometry-Based Secretome Analysis as a Tool to Investigate Metalloprotease and TIMP Activity. Methods Mol. Biol. 2020, 2043, 265–273.
- Scilabra, S.D.; Pigoni, M.; Pravata, V.; Schatzl, T.; Muller, S.A.; Troeberg, L.; Lichtenthaler, S.F. Increased TIMP-3 expression alters the cellular secretome through dual inhibition of the metalloprotease ADAM10 and ligand-binding of the LRP-1 receptor. Sci. Rep. 2018, 8, 14697.
- Carreca, A.P.; Pravata, V.M.; D’Apolito, D.; Bonelli, S.; Calligaris, M.; Monaca, E.; Muller, S.A.; Lichtenthaler, S.F.; Scilabra, S.D. Quantitative Proteomics Reveals Changes Induced by TIMP-3 on Cell Membrane Composition and Novel Metalloprotease Substrates. Int. J. Mol. Sci. 2021, 22, 2392.
- Fan, D.; Kassiri, Z. Biology of Tissue Inhibitor of Metalloproteinase 3 (TIMP3), and Its Therapeutic Implications in Cardiovascular Pathology. Front. Physiol. 2020, 11, 661.
- Tushaus, J.; Muller, S.A.; Kataka, E.S.; Zaucha, J.; Sebastian Monasor, L.; Su, M.; Guner, G.; Jocher, G.; Tahirovic, S.; Frishman, D.; et al. An optimized quantitative proteomics method establishes the cell type-resolved mouse brain secretome. EMBO J. 2020, 39, e105693.
- Kuhn, P.H.; Koroniak, K.; Hogl, S.; Colombo, A.; Zeitschel, U.; Willem, M.; Volbracht, C.; Schepers, U.; Imhof, A.; Hoffmeister, A.; et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012, 31, 3157–3168.
- Herber, J.; Njavro, J.; Feederle, R.; Schepers, U.; Muller, U.C.; Brase, S.; Muller, S.A.; Lichtenthaler, S.F. Click Chemistry-mediated Biotinylation Reveals a Function for the Protease BACE1 in Modulating the Neuronal Surface Glycoproteome. Mol. Cell. Proteom. 2018, 17, 1487–1501.
- Prudova, A.; Serrano, K.; Eckhard, U.; Fortelny, N.; Devine, D.V.; Overall, C.M. TAILS N-terminomics of human platelets reveals pervasive metalloproteinase-dependent proteolytic processing in storage. Blood 2014, 124, e49–e60.
- Doucet, A.; Kleifeld, O.; Kizhakkedathu, J.N.; Overall, C.M. Identification of proteolytic products and natural protein N-termini by Terminal Amine Isotopic Labeling of Substrates (TAILS). Methods Mol. Biol. 2011, 753, 273–287.
- Das, N.; Benko, C.; Gill, S.E.; Dufour, A. The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors. Pharmaceuticals 2020, 14, 31.
- Anand-Apte, B.; Chao, J.R.; Singh, R.; Stohr, H. Sorsby fundus dystrophy: Insights from the past and looking to the future. J. Neurosci. Res. 2019, 97, 88–97.
- Lobb, D.C.; Doviak, H.; Brower, G.L.; Romito, E.; O’Neill, J.W.; Smith, S.; Shuman, J.A.; Freels, P.D.; Zellars, K.N.; Freeburg, L.A.; et al. Targeted Injection of a Truncated Form of Tissue Inhibitor of Metalloproteinase 3 Alters Post-Myocardial Infarction Remodeling. J. Pharmacol. Exp. Ther. 2020, 375, 296–307.